一种二氢吡啶类药物的口服固体制剂及其制备方法与流程
:
1.本技术属于药物制剂技术领域,具体涉及一种二氢吡啶类药物非洛地平的口服固体制剂及其制备方法。
背景技术:
2.二氢吡啶是钙拮抗剂中的重要药物,主要包括硝苯地平、尼莫地平、尼群地平、苯环酸氨氯地平、拉西地平、尼卡地平、非洛地平、西尼地平等。
3.以非洛地平((
±
)-2,6-二甲基-4-(2,3-二氯苯基)-1,4-二氢-3,5-吡啶二甲酸甲酯乙酯)为例,其具有以下通式ⅰ所示的化合物:
[0004][0005]
非洛地平为bcsⅱ类水溶性差而渗透性好的药物,其在水中溶解度为0.00058mg/ml,因此提高非洛地平溶出为制备非洛地平制剂的重点和难点。
[0006]
专利cn102462663a公开了一种降压药非洛地平药物组合物及其制备方法,该组合物包括非洛地平、填充剂、分散剂、崩解剂、增溶剂、粘合剂、润滑剂,方法是先将非洛地平与分散剂一起微粉化,再采用干法制粒制备非洛地平药物组合物,其中使用了甘露醇作为增溶剂,而原研制剂没使用增溶剂。
[0007]
专利cn102462663a公开了一种非洛地平药物固体分散体及其制备方法,其中难溶性药物非洛地平为活性成分,将活性药物与高分子载体按照一定比例混匀,通过热熔挤出法制得非洛地平固体分散体。
[0008]
专利cn104758938a公开了一种治疗高血压的非洛地平片剂及其制备方法,该片剂含有非洛地平、吐温80、交联聚维酮和填充剂。制备方法为:将非洛地平溶解在乙醇中,然后加入吐温80、交联聚维酮,搅拌均匀,以此混悬液作为粘合剂在填充剂上制粒、干燥得到含药颗粒,将此含药颗粒与润滑剂混合均匀,压片而得。
[0009]
上述现有技术方法在一定程度上对非洛地平药物的溶解起到了增溶效果,但均存在不同程度的导致药物不稳定的因素,专利文献1为了增加药物溶出,使用了增溶剂甘露醇;专利文献2为了增加药物溶出,将药物制备成固体分散体,容易形成不稳定晶型,不利于药物稳定性,且固体分散体技术工艺较复杂,对生产设备和操作的要求较高;专利文献3将非洛地平溶解在乙醇中,且使用表面活性剂吐温80,不仅容易使非洛地平形成不稳定晶型,还会导致体内外溶出行为不一致。
技术实现要素:
[0010]
本发明的目的在于提供一种不含增溶剂、不含表面活性剂,制剂工艺简单、溶出迅速、长期稳定的二氢吡啶类药物口服固体制剂。
[0011]
本发明的另一目的在于提供一种制备上述口服制剂的方法。
[0012]
一种二氢吡啶类药物口服固体制剂,包含d50为0.1~28μm的二氢吡啶类药物、水溶性多羟基化合物、非水溶性填充剂、水溶性聚合物粘合剂。
[0013]
研究发现,在所述的粒径原料药以及成分控制下,能够改善制剂的溶出性能。
[0014]
本发明研究发现,在所述的成分联合下,进一步对二氢吡啶类药物的粒径范围进行控制,可以进一步实现协同,进一步改善溶出效果。
[0015]
作为优选,所述的二氢吡啶类药物d50为0.5~20μm;进一步优选为1~10μm,更进一步优选为2~5μm。
[0016]
本发明研究发现,在优选的粒径下,进一步配合成分的联合,能够进一步实现协同,可进一步改善二氢吡啶类药物制剂溶出效果。
[0017]
本发明中,所述的二氢吡啶类药物为非洛地平、硝苯地平、尼群地平、西尼地平中的至少一种。
[0018]
优选地,所述的水溶性多羟基化合物为乳糖、山梨醇或葡萄糖中的一种或多种。
[0019]
作为优选,所述的水溶性多羟基化合物为乳糖,还包含山梨醇、葡萄糖中的至少一种。
[0020]
优选地,水溶性多羟基化合物中,乳糖的重量百分数为不低于40wt.%;进一步优选为50~80wt.%。
[0021]
本发明中,非水溶性填充剂为微晶纤维素、玉米淀粉、糊精、聚乙烯吡咯烷酮、蔗糖或甘露醇中的一种或多种;优选地,所述的非水溶性填充剂为微晶纤维素和玉米淀粉;优选地,二者的重量比为1:1~2。
[0022]
优选地,水溶性聚合物粘合剂为羟丙纤维素、羟丙甲纤维素中的至少一种。
[0023]
本发明中,所述的固体制剂还可根据制剂需要添加其他辅助成分,例如,所述的辅助成分包括但不限于润滑剂、着色剂、矫味剂等中的至少一种;
[0024]
其中,润滑剂可以是行业内公知的润滑成分,例如硬脂酸镁。
[0025]
着色剂为行业内公知的着色成分,例如,可以是氧化铁、二氧化钛等。
[0026]
矫味剂为常见矫味剂,例如,可以是阿司帕坦、薄荷醇、蔗糖、果糖、甜叶菊素、水果味香精等。
[0027]
所述的二氢吡啶类药物口服固体制剂中,二氢吡啶类药物的含量为1~10wt.%、非水溶性填充剂的含量为20~40wt.%;水溶性聚合物粘合剂的含量为0.1~1wt.%;余量为水溶性多羟基化合物;各成分总含量为100wt.%;
[0028]
优选地,所述的二氢吡啶类药物口服固体制剂中,辅助成分的含量为0.5~2.5wt.%;
[0029]
进一步优选,二氢吡啶类药物口服固体制剂中,二氢吡啶类药物的含量为3~7wt.%、非水溶性填充剂的含量为30~35wt.%;水溶性聚合物粘合剂的含量为0.1~0.4wt.%;辅助成分的含量为1~1.5wt.%;余量为水溶性多羟基化合物;各成分总含量为100wt.%。
[0030]
本发明技术方案的另一实施方案:所述的水溶性多羟基化合物包覆在二氢吡啶类药物表面构建成核-壳结构颗粒。
[0031]
本发明中,通过水溶性多羟基化合物壳对二氢吡啶类药物核进行包覆,进一步通过二氢吡啶类药物核粒径的联合控制,能够实现协同,能够有效改善二氢吡啶类药物的溶出性能以及稳定性。
[0032]
本发明研究发现,所述的核的粒径以及核-壳结构的联合协同是进一步改善溶出性以及稳定性的关键。本发明研究还发现,进一步对核以及壳的成分以及颗粒级配,有助于进一步实现协同,更进一步改善二氢吡啶类药物的溶出性能以及稳定性。
[0033]
作为优选,所述的核包括二氢吡啶类药物颗粒a和二氢吡啶类药物颗粒b;
[0034]
其中,二氢吡啶类药物颗粒a的d50为0.5~6μm,进一步优选为2~3μm;二氢吡啶类药物颗粒b的d50为15~28μm。进一步优选为20~26μm。本发明研究发现,采用所述粒径级配的核的联合,能够进一步实现协同,进一步改善二氢吡啶类药物的溶出性能以及稳定性。
[0035]
优选地,二氢吡啶类药物颗粒a和二氢吡啶类药物颗粒b的重量比为1:0.5~2;进一步优选为1:1~1.5。
[0036]
作为优选,所述的核中,还包括水溶性纤维c。研究发现,在所述的核的级配下,进一步配合所述的微观纤维状的水溶性纤维c,能够意外地进一步构建三维溶出通道,能够意外地进一步协同改善溶出性以及稳定性。
[0037]
优选地,所述的水溶性纤维c为羟丙纤维素、羟丙甲纤维素中的至少一种。
[0038]
优选地,所述的水溶性纤维c在所述的固体制剂中的重量占比不高于1wt.%;优选为0.1~0.3wt.%。
[0039]
本发明中,采用所述的水溶性多羟基化合物作为壳。作为优选,所述的水溶性多羟基化合物为乳糖,还包含山梨醇、葡萄糖中的至少一种。优选地,壳中,乳糖的重量百分数不低于(大于或等于)40wt.%;进一步优选为50~80wt.%。进一步优选,所述的壳中包括乳糖和山梨醇,且二者的重量比例为1~3:1;进一步优选为1.5~2:1。研究发现,在所述的核-壳、级配的核以及组合的壳的结构和成分联合下,能够进一步协同,进一步改善溶出效果。
[0040]
本发明优选的二氢吡啶类药物口服固体制剂中,核的含量为3~7wt.%、非水溶性填充剂的含量为30~35wt.%;水溶性聚合物粘合剂的含量为0.1~0.4wt.%;辅助成分的含量为1~1.5wt.%;余量为壳;各成分总含量为100wt.%。
[0041]
本发明优选的方案,通过所述的核-壳结构、核级配以及壳成分联合控制,能够实现协同,能够改善二氢吡啶类药物的溶出,可以在不添加增溶剂、不含表面活性剂下,即可获得良好的溶出性以及稳定性。此外,该制剂还具有优异的稳定性。
[0042]
本发明中,所述的口服固体制剂,为片剂、颗粒剂、胶囊剂等中的至少一种。
[0043]
本发明还提供了一种所述的二氢吡啶类药物口服固体制剂的制备方法,将各成分混合,制得所述的二氢吡啶类药物口服固体制剂。
[0044]
例如,所述的制备为:将二氢吡啶类药物微粒、水溶性多羟基化合物、非水溶性填充剂混合均匀,加入水溶性聚合物粘合剂制粒,随后和其他成分(如辅助成分)混合,制得所述的二氢吡啶类药物口服固体制剂。
[0045]
本发明优选的包覆结构的二氢吡啶类药物口服固体制剂,其制备方法为:预先将水溶性多羟基化合物包覆在二氢吡啶类药物表面构建成核-壳结构颗粒,随后和其他成分
混合,制得所述的二氢吡啶类药物口服固体制剂;
[0046]
进一步优选,二氢吡啶类药物颗粒a和二氢吡啶类药物颗粒b混合成核,随后在其表面包覆水溶性多羟基化合物,形成核-壳结构颗粒,随后和其他成分混合,制得所述的二氢吡啶类药物口服固体制剂;
[0047]
更进一步优选,将二氢吡啶类药物颗粒a、二氢吡啶类药物颗粒b、水溶性纤维c混合成核,随后在其表面包覆水溶性多羟基化合物,形成核-壳结构颗粒,随后和其他成分混合,制得所述的二氢吡啶类药物口服固体制剂。
[0048]
以非洛地平口服制剂为例,其制备过程包括:
[0049]
(1)获得需要的非洛地平颗粒;
[0050]
(2)将非洛地平微粒级配或选择性和水溶性纤维c混合,随后与水溶性多羟基化合物置于湿法制粒机中,使水溶性多羟基化合物将非洛地平微粒充分包覆,形成核-壳颗粒。
[0051]
(4)制粒:向制粒机中加入非水溶性填充剂、核-壳颗粒混合均匀,加入水溶性聚合物粘合剂溶液制粒。
[0052]
(5)干燥:将步骤(4)制得的颗粒干燥,控制水分在3wt%以内,优选为0.5~1.5wt%。
[0053]
(6)整粒:干燥后的颗粒采用筛网整粒。
[0054]
(7)压片:将所得颗粒与处方量的硬脂酸镁混匀,压片,制备的片剂片重差异符合要求(片重差异小于7.5%)。
[0055]
有益效果
[0056]
1、研究发现,在所述的粒径原料药以及成分控制下,能够改善制剂的溶出性能。进一步通过所述的成分以及粒径的联合控制,有助于进一步改善制剂的溶出性能,降低制剂中的杂质产生,提高药物的安全性。
[0057]
2、本发明所述的制剂不仅在体外的溶出与参比制剂一致,且口服吸收良好,具有较好的生物利用度。3、本发明所述的制剂,制备工艺简单可行,制得的制剂质量稳定可控,利于工业大规模生产。
附图说明
[0058]
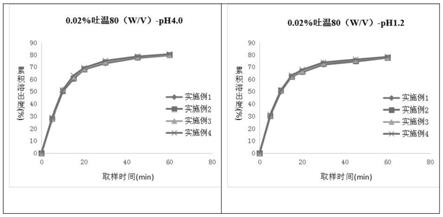
附图1为实施例1~4的溶出曲线,根据累积溶出度数据拟合得到。
[0059]
附图2为实施例5~14的溶出曲线,根据累积溶出度数据拟合得到。
具体实施方案
[0060]
非洛地平颗粒粒径控制:采用yq-200型气流粉碎机(由上海赛山粉体机械制造有限公司制造)进行粒径控制,条件:进料压力为0.8mpa,粉碎压力为0.8mpa,进料速度为5hz。
[0061]
非洛地平平均粒径测定:取粉碎后的非洛地平适量,加入适量水搅拌,再加入适量吐温-80,超声使非洛地平分散均匀,采用3000e型激光粒度分析仪(malvern公司生产)测定。
[0062]
样品制备采用湿法制粒工艺制备样品,设备为g10型湿法混合制粒机(由浙江迦南科技股份有限公司生产制造),制粒锅1l。
[0063]
溶出试验可较好地模拟口服固体制剂在消化道中的释放行为,是一种模拟口服固
体制剂在胃肠道中的崩解和溶出的体外试验方法,它是评价药物制剂质量和日常监管的一个重要指标,也是制剂研发筛选处方的一个重要工具。本品按《中国药典》2020版通则0931溶出度与释放度测定法第二法(桨法)进行溶出试验。试验溶出液参考日本橙皮书《非洛地平片5mg溶出试验》所用溶出液配制,为0.02%吐温80(w/v)-ph4.0溶液和0.02%吐温80(w/v)-ph1.2溶液,50rpm,试验液:900ml。
[0064]
实施例1
[0065]
处方:
[0066]
非洛地平微粒(平均粒径:20μm)15g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g硬脂酸镁3.0g
[0067]
样品制备:将非洛地平微粒、微晶纤维素、玉米淀粉、乳糖加入湿法制粒机中,开启搅拌功能和剪切功能,使混合均匀,加入浓度为3%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用筛网整粒,将其与处方量的硬脂酸镁混匀,使用旋转式压片机制备成非洛地平含量为5mg的片剂,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0068]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表1。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0069]
实施例2
[0070]
处方:
[0071]
非洛地平微粒(平均粒径:10μm)15g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g硬脂酸镁3.0g
[0072]
和实施例1相比,区别仅在于,非洛地平的d50为10μm,其他参数均同实施例1。
[0073]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表1。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0074]
实施例3
[0075]
处方:
[0076]
非洛地平微粒(平均粒径:5μm)15g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g
硬脂酸镁3.0g
[0077]
和实施例1相比,区别仅在于,非洛地平的d50为5μm,其他参数均同实施例1。
[0078]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表1。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0079]
实施例4
[0080]
处方:
[0081]
非洛地平微粒(平均粒径:2.3μm)15g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g硬脂酸镁3.0g
[0082]
和实施例1相比,区别仅在于,非洛地平的d50为2.3μm,其他参数均同实施例1。
[0083]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表1。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0084]
实施例5
[0085]
处方:
[0086]
非洛地平微粒(平均粒径:2.3μm)15g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g硬脂酸镁3.0g
[0087]
和实施例4相比,处方相同,区别仅在于,预先形成核壳结构:
[0088]
样品制备:将非洛地平微粒(核)、乳糖(壳)于湿法制粒机中,开启搅拌功能和剪切功能,使乳糖将非洛地平微粒充分包覆,形成包覆共同体。将包覆共同体与微晶纤维素、玉米淀粉加入湿法制粒机混合均匀,加入浓度为3%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用φ0.8mm筛网整粒,将其与处方量的硬脂酸镁混匀,压片,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0089]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0090]
实施例6
[0091]
和实施例5相比,区别仅在于,非洛地平颗粒的d50为26μm。
[0092]
处方:
[0093]
非洛地平微粒(d50:26.0μm)15g乳糖163g微晶纤维素32g玉米淀粉56g
羟丙纤维素1.0g硬脂酸镁3.0g
[0094]
样品制备:将非洛地平微粒(核)、乳糖(壳)于湿法制粒机中,开启搅拌功能和剪切功能,使乳糖将非洛地平微粒充分包覆,形成包覆共同体。将包覆共同体与微晶纤维素、玉米淀粉加入湿法制粒机混合均匀,加入浓度为3%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用筛网整粒,将其与处方量的硬脂酸镁混匀,使用旋转式压片机制备成非洛地平含量为5mg的片剂,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0095]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0096]
实施例7
[0097]
和实施例5相比,区别仅在于,非洛地平颗粒包括颗粒a和b,其中,颗粒a的d50为2.3μm,颗粒b的d50为26μm。颗粒a和b的质量比为1:1。
[0098]
处方:
[0099]
非洛地平颗粒a(d50:2.3μm)7.5g非洛地平颗粒b(d50:26.0μm)7.5g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g硬脂酸镁3.0g
[0100]
样品制备:将非洛地平颗粒a(核)、非洛地平颗粒b(核)预先混合,随后和乳糖(壳)于湿法制粒机中,开启搅拌功能和剪切功能,使乳糖将非洛地平微粒充分包覆,形成包覆共同体。将包覆共同体与微晶纤维素、玉米淀粉加入湿法制粒机混合均匀,加入浓度为3%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用筛网整粒,将其与处方量的硬脂酸镁混匀,使用旋转式压片机制备成非洛地平含量为5mg的片剂,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0101]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0102]
实施例8
[0103]
和实施例7相比,区别仅在于,非洛地平颗粒包括颗粒a和b,其中,颗粒a的d50为2.3μm,颗粒b的d50为26μm。颗粒a和b的质量比为1:2。
[0104]
处方:
[0105][0106][0107]
样品制备:将非洛地平颗粒a(核)、非洛地平颗粒b(核)预先混合,随后和乳糖(壳)于湿法制粒机中,开启搅拌功能和剪切功能,使乳糖将非洛地平微粒充分包覆,形成包覆共同体。将包覆共同体与微晶纤维素、玉米淀粉加入湿法制粒机混合均匀,加入浓度为3%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用筛网整粒,将其与处方量的硬脂酸镁混匀,使用旋转式压片机制备成非洛地平含量为5mg的片剂,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0108]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0109]
实施例9
[0110]
和实施例7相比,区别仅在于,非洛地平颗粒包括颗粒a和b,其中,颗粒a的d50为5.2μm,颗粒b的d50为21.0μm。颗粒a和b的质量比为1:1。
[0111]
处方:
[0112]
非洛地平颗粒a(d50:5.2μm)7.5g非洛地平颗粒b(d50:21.0μm)7.5g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素1.0g硬脂酸镁3.0g
[0113]
样品制备:将非洛地平颗粒a(核)、非洛地平颗粒b(核)预先混合,随后再和乳糖(壳)于湿法制粒机中,开启搅拌功能和剪切功能,使乳糖将非洛地平微粒充分包覆,形成包覆共同体。将包覆共同体与微晶纤维素、玉米淀粉加入湿法制粒机混合均匀,加入浓度为3%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用筛网整粒,将其与处方量的硬脂酸镁混匀,使用旋转式压片机制备成非洛地平含量为5mg的片剂,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0114]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0115]
实施例10
[0116]
和实施例7相比,区别仅在于,核中还添加有水溶性纤维聚合物c(羟丙纤维素),其中,水溶性纤维聚合物c相对于处方中羟丙纤维素总量的50%。也即是,处方中的羟丙纤维素50%预先固体混合至核中,剩余部分作为水溶性粘合剂使用。
[0117]
样品制备:将非洛地平微粒a+b(核)预先和羟丙纤维素混合,随后再和乳糖(壳)于湿法制粒机中,开启搅拌功能和剪切功能,使乳糖将非洛地平微粒充分包覆,形成包覆共同体。将包覆共同体与微晶纤维素、玉米淀粉加入湿法制粒机混合均匀,加入浓度为1.5%(wt/wt)羟丙纤维素水溶液制粒,干燥上述含药颗粒,然后采用筛网整粒,将其与处方量的硬脂酸镁混匀,得制剂粉料,使用旋转式压片机制备成非洛地平含量为5mg的片剂,压片过程顺利,制备的片剂片重差异符合规定(片重差异小于7.5%)。
[0118]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0119]
实施例11
[0120]
和实施例10相比,区别仅在于,采用山梨醇替换所述的乳糖作为壳,其他参数均同实施例10。
[0121]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0122]
实施例12
[0123]
和实施例10相比,区别仅在于,采用质量比为1:1的乳糖和山梨醇的混合物(处方中水溶性多羟基化合物的总含量为163g)替换所述的乳糖作为壳,其他参数均同实施例10。
[0124]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0125]
实施例13
[0126]
和实施例12相比,区别仅在于,采用质量比为2:1的乳糖和山梨醇的混合物作为壳,其他参数同实施例12。
[0127]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0128]
实施例14
[0129]
和实施例13相比,区别在于,采用质量比为2:1的乳糖和葡萄糖的混合物作为壳,其他参数同实施例13。
[0130]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0131]
实施例15
[0132]
和实施例13相比,区别仅在于,采用糊精替换(微晶纤维素-玉米淀粉)作为非水溶性填充剂。其他参数同实施例13。
[0133]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0134]
实施例16
[0135]
和实施例13相比,区别仅在于,将处方制备胶囊剂,步骤为:
[0136]
样品制备:将制剂粉料采用3号胶囊壳灌装成非洛地平含量为5mg/粒的胶囊,灌装
过程顺利。
[0137]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表2。从溶出结果可知,样品在上述2个介质中为快速溶出。
[0138]
对比例1
[0139]
和实施例1相比,区别仅在于,核的d50粒径为32.0μm,其他操作均同实施例1。
[0140]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表3。从溶出结果可知,样品在上述2个介质中溶出较慢,未达到快速溶出的目的。
[0141]
对比例2:
[0142]
和实施例1相比,区别仅在于,核的d50粒径为0.06μm,其他操作均同实施例1。
[0143]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表3。从溶出结果可知,样品在上述2个介质中溶出较慢,未达到快速溶出的目的。
[0144]
对比例3
[0145]
和实施例1相比,区别仅在于,粘合剂羟丙纤维素超出本发明范围,
[0146]
处方:
[0147]
非洛地平微粒15g乳糖163g微晶纤维素32g玉米淀粉56g羟丙纤维素4.0g硬脂酸镁3.0g
[0148]
测定样品在0.02%吐温80-ph4.0介质和0.02%吐温80-ph1.2介质中的溶出曲线,结果见表3。从溶出结果可知,样品在上述2个介质中溶出较慢,未达到快速溶出的目的。
[0149]
三、效果数据
[0150]
1、溶出数据
[0151]
取各实施例下样品测定溶出曲线:以0.02%吐温80(w/v)-ph4.0为溶出介质1和以0.02%吐温80(w/v)-ph1.2为溶出介质2;桨法,转速为每分钟50转,设置5、10、15、20、30、45、60、90min溶出曲线测定取样时间点,于各时间点取样,并即时补充相同温度、相同体积的溶出介质,滤过,取续滤液,采用高效液相色谱法测定溶出度。根据溶出度计算累积溶出度、绘制溶出曲线,结果如下:
[0152]
表1实施例1~4的溶出曲线测定结果表
[0153][0154]
表2.实施例5~16的溶出曲线测定结果表
[0155][0156][0157]
表3.对比例1~3的溶出曲线测定结果表
[0158][0159]
试验结论:
[0160]
由实施例1~4实验数据可以看出,api粒径大小对样品在0.02%吐温80(w/v)-ph4.0介质和0.02%吐温80(w/v)-ph1.2介质中的溶出产生影响,api粒径越小,溶出越大。
[0161]
由实施例5~10实验数据可以看出,核壳结构中,单一粒径的原料药粒径大小对溶出有明显影响(实施例5、6),粒径越小溶出越快;不同粒径的原料药按一定比例进行组合比单一粒径原料制备的样品溶出明显提高(实施例5~9),其中最优的粒径组合物为颗粒a(d50=2.3μm):颗粒b(d50=26μm)=1:1(实施例7);在核中添加水溶性纤维聚合物c可进一步提高溶出(实施例10)。
[0162]
实施例10~14主要对包覆共同体(核-壳结构)的壳进行筛选,考察了乳糖、山梨醇及葡萄糖作为壳对样品溶出的影响,考察结果为不同壳对溶出有明显影响,其中最优的壳的组合物为乳糖:山梨醇=2:1(实施例13)。
[0163]
2.稳定性考察
[0164]
按照《中国药典》2020版9001“原料药物与制剂稳定性试验指导原则”考察含非洛地平制剂的稳定性。
[0165]
按照本发明实施例的方法制备一批样品,进行稳定性考察,样品放置条件及考察项目见下表。
[0166]
表4考察项目及条件
[0167][0168]
稳定性试验结果见下表:
[0169]
表5.稳定性试验数据
[0170]
[0171][0172]
试验结论:表中各实施例稳定性数据均符合要求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1