一种固有无序蛋白质预测方法
1.本发明属于生物信息学领域,涉及一种使用新颖特征的固有无序蛋白质预测方案。
背景技术:
2.蛋白质中没有稳定三维结构的区域被称为蛋白质的无序区域,包含至少一段无序区域的蛋白质被称为固有无序蛋白质。由于灵活的结构使得其广泛参与了重要的生理过程,如核酸折叠、dna转录和翻译、分子识别和分子组装以及蛋白质之间的相互作用等。另外很多人类的疾病,如癌症、阿兹海默症和一些遗传疾病等都与其相关。因此快速且准确的预测固有无序蛋白质对蛋白质功能的理解、药物研发以及一些疾病的治疗等方面有着重要的意义。在过去,有很多实验方法用来测定蛋白质的无序区域,但是实验方法往往成本高且耗时长,不能进行大规模的测定。因此通过计算方法对固有无序蛋白质进行预测是一个简单高效的方法。
技术实现要素:
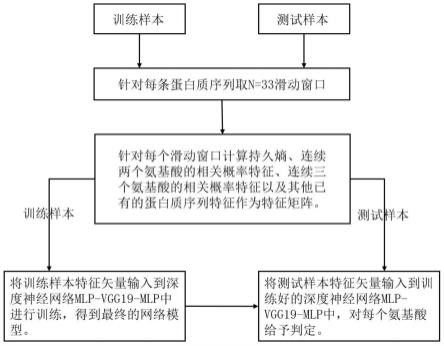
3.本发明的目的是克服现有技术存在的上述不足,设计一种固有无序蛋白质预测方法,该方法使用三个新颖的序列特征即持久熵、连续两个氨基酸的相关概率特征和连续三个氨基酸的相关概率特征以及其他已有的蛋白质序列特征作为特征矩阵,利用vgg199变体和两个mlp网络构建深度神经网络实现了较准确的固有无序蛋白质的预测。
4.本发明提供的固有无序蛋白质预测方法的具体步骤如下:
5.第1、针对学习样本,令w表示其中一条长度为l蛋白质序列,用长度为n的滑动窗口截取相应长度的连续氨基酸片段βj=wj…wj+(n-1)
进行计算;
6.第2、首先计算持久熵,公式如下:
[0007][0008]
其中
[0009][0010]
其中δ(
·
)是冲激函数,用一一对应的顺序表示氨基酸符号集为:
[0011][0012]
则持久熵为:
[0013][0014]
其中表示与条形码图(假定对于所有的1≤i≤n)相关的滤流,它的k维条形码[εs,εe)表示滤流在时刻εs出现在时刻εe结束。
[0015]
第3、计算连续两个和三个氨基酸的相关概率特征,公式如下:
[0016]
定义两个集合:
[0017][0018][0019]
表示在蛋白质序列中连续两个和三个氨基酸的所有的可能性。则两个新的特征为:
[0020]
h2=[h2(1),
…
,h2(l)]
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(7)
[0021]
h3=[h3(1),...,h3(l)]
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(8)
[0022]
其中
[0023][0024][0025]
式中函数i2(j)和i3(j)的定义为:
[0026][0027][0028]
其中wj和分别表示wjw
j+1
(1≤j≤l-1)和wjw
j+1wj+2
(1≤j≤l-2);
[0029]
[0030][0031]
其中,和
[0032][0033][0034]
上式中
[0035][0036][0037]
其中,
[0038][0039][0040]
其中wj和分别表示wjw
j+1
(1≤j≤l-1)和wjw
j+1wj+2
(1≤j≤l-2)。
[0041]
第4、针对一条长度为l的蛋白质序列w,计算该序列的20种进化信息、7种物理化学性质、3种倾向性以及香浓熵和拓扑熵。联合上述三个新颖的特征序列得到最终的特征矩阵。
[0042]
第5、利用十折交叉验证训练深度神经网络,得到最终的网络参数。
[0043]
第6、对于待预测的蛋白质序列,按照步骤第1至第4步计算各个残基的特征矢量,利用第5步得到的神经网络参数进行氨基酸的判定。
[0044]
本发明的优点和积极效果:
[0045]
该方案利用三个新颖的序列特征即持久熵、连续两个氨基酸的相关概率特征和连续三个氨基酸的相关概率特征以及其他已有的蛋白质序列特征作为特征矩阵。三个新颖的序列特征是第一次被提出用于固有无序蛋白质的预测;其次,该方法利用vgg199变体和两个mlp网络构建深度神经网络mlp-vgg19-mlp实现了较准确的固有无序蛋白质的预测。
附图说明
[0046]
图1是:实现本发明预测固有无序蛋白质方法的流程图。
具体实施方式
[0047]
参见附图1,本发明使用三个新颖的序列特征即持久熵、连续两个氨基酸的相关概
率特征和连续三个氨基酸的相关概率特征以及其他已有的蛋白质序列特征作为特征矩阵,利用vgg199变体和两个mlp网络构建深度神经网络实现了较准确的固有无序蛋白质的预测;
[0048]
其中包括针对学习样本蛋白质序列,用长度为n的滑动窗口截取相应长度的连续氨基酸片段的步骤;
[0049]
包括针对每个滑动窗口计算持久熵、连续两个氨基酸的相关概率特征、连续三个氨基酸的相关概率特征以及其他已有的蛋白质序列特征作为特征矩阵的步骤;
[0050]
将训练样本特征矢量输入到深度神经网络mlp-vgg19-mlp中进行训练,得到最终网络模型的步骤;
[0051]
将测试样本特征输入到训练好的深度神经网络mlp-vgg19-mlp中,对每个氨基酸给予判定的步骤。
[0052]
下面结合实施例对本发明实现方式做进一步说明。
[0053]
实施例1:
[0054]
本发明提供的固有无序蛋白质的预测方法具体步骤如下:
[0055]
针对一条未判定无序区域的蛋白质序列w(以r80数据集中一条标号为1lon的蛋白
[0056]
质序列为例),利用本发明提供的固有无序蛋白质预测方案进行预测的具体步骤如下:
[0057]
步骤一:该序列长度为457,用n=33的滑动窗口对序列进行截取。针对每个窗口区间计算三十五种特征的值。
[0058]
序列w=gtrasndrppgtggvkrgrlqqeaaatgsrvtv
[0059]
针对第一个长度为n的滑动窗口,通过公式(4)(7)(8)计算持久熵、连续两个氨基酸的相关概率特征和连续三个氨基酸的相关概率特征值,同时计算20种进化信息、7种物理化学性质、3种倾向性以及香浓熵和拓扑熵特征值共获得35种特征值,之后滑动窗口计算对应的特征值得到最终的特征矩阵。
[0060]
计算得到的蛋白质序列w的特征矩阵如下,其中每一列对应该位置氨基酸的特征矢量:
[0061][0062]
步骤二:利用学习样本训练深度神经网络mlp-vgg19-mlp得到的最终的网络参数对x进行判定。
[0063]
为了验证该预测方法的有效性,利用dis166数据集、r80数据集和mxd494数据集对该方法进行了固有无序蛋白质的预测。其中,dis166数据集包含166条蛋白质序列;r80数据集中包含80条蛋白质序列,mxd494数据集中包含494条蛋白质序列。表1中列出了针对dis166数据集,本发明设计的固有无序蛋白质的预测方法与现有的同类型预测方法的预测准确度比较。表2列出了针对r80数据集,本发明设计的天然无序蛋白质的预测方法与现有的同类型预测方法的预测准确度比较。表3列出了针对r494数据集,本发明设计的天然无序
of intrinsically disordered proteins in plant signaling[j].biochemical journal,2012,442(1):1-12.
[0076]
4、yang z r,thomson r,mcneil p,et al.ronn:the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins[j].bioinformatics,2005,21(16):3369-3376.
[0077]
5、comprehensive comparative assessment of in-silico predictors of disordered regions[j].current protein and peptide science,2012,13(1):6-18.
[0078]
6、hatos a,hajdu-solt
é
sz b,monzon a m,et al.disprot:intrinsic protein disorder annotation in 2020[j].nucleic acids research,2020,48(d1):d269-d276.
[0079]
7、he h,zhao j.a low computational complexity scheme for the prediction of intrinsically disordered protein regions[j].mathematical problems in engineering,2018,2018.
[0080]
8、meiler j,m
ü
ller m,zeidler a,et al.generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks[j].molecular modeling annual,2001,7(9):360-369.
[0081]
9、xu p,zhao j,zhang j.identification of intrinsically disordered protein regions based on deep neural network-vgg16[j].algorithms,2021,14(4):107.
[0082]
10、hanson j,paliwal k k,litfin t,et al.spot-disorder2:improved protein intrinsic disorder prediction by ensembled deep learning[j].genomics,proteomics&bioinformatics,2019,17(6):645-656.
[0083]
11、liu y,wang x,liu b.rfpr-idp:reduce the false positive rates for intrinsically disordered protein and region prediction by incorporating both fully ordered proteins and disordered proteins[j].briefings in bioinformatics,2021,22(2):2000-2011.
[0084]
12、tang y j,pang y h,liu b.idp-seq2seq:identification of intrinsically disordered regions based on sequence to sequence learning[j].bioinformatics,2020,36(21):5177-5186.
[0085]
13、he h,zhao j,sun g.the prediction of intrinsically disordered proteins based on feature selection[j].algorithms,2019,12(2):46。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1