与癌症相关的遗传或分子畸变的检测
与癌症相关的遗传或分子畸变的检测
1.相关申请的交叉引用
2.本技术要求2010年11月30日提交的题为“detection of genetic aberrations associated with cancer”的美国临时申请no.61/418,391以及2011年8月31日提交的题为“detection of genetic or molecular aberrations associated with cancer”的美国临时申请no.61/529,877的优先权,并且为它们的非临时申请,就所有目的而言,这些申请的全部内容以引用方式并入本文。
3.本技术涉及2010年11月5日提交的题为“size-based genomic analysis”的共同拥有的美国专利申请no.12/940,992(美国公开2011/0276277)(lo et al.)(代理人案卷号80015-794101/006610us)以及2010年11月5日提交的题为“fetal genomic analysis from a maternal biological sample”的美国专利申请no.12/940,993(美国公开2011/0105353)(lo et al.)(代理人案卷号80015-794103/006710us),这些申请的公开内容以引用方式全部并入本文。
背景技术:
4.癌症是影响许多人的常见疾病。其通常直到严重症状出现时才会被发现。就常见类型的癌症而言,目前虽然已存在一些癌症筛查技术来判断患者患有癌症的可能性,但是此类癌症筛查技术的可靠性和准确性通常是比较低,或者其需要让患者接受较高剂量的辐射。就许多其他类型的癌症而言,目前尚未存在有效的筛查技术。
5.在肺癌以及头颈癌的患者的外周血dna中,检测到特定基因座的杂合性缺失(loh)(chen xq,et al.nat med 1996;2:1033-5;nawroz h,et al.nat med 1996;2:1035-7)。但是,可以检测到的loh相对量较少事实阻碍了此类技术用于检测特定的基因座。即使在使用数字pcr,这些方法仍不能检测到相对量较少的loh。此外,此类技术仍局限于研究在特定癌症类型中发生的已知的特定基因座。因此,不能或不能有效地将此类方法作为通用的手段去筛查各种癌症。
6.除了筛查癌症的存在与否存在局限性以外,目前的技术更不能有效地为癌症患者提供预后诊断以及治疗效果的监测(例如在手术、化疗或免疫治疗或靶向治疗之后的恢复)。因为此类技术通常是价格昂贵(例如成像技术)、准确度低、效率低、敏感度低,并且如果应用成像技术,可能使患者受到辐射的影响。
7.因此,理想的发明是能够有效地为癌症患者提供筛查、预后和监测的新技术。
8.发明概述
9.本发明提供了多个实施方案用于检测与癌症相关的遗传畸变的系统、仪器和方法。对包括游离dna片段的生物样品进行分析,从而鉴别肿瘤中例如由于染色体片段缺失和/或扩增而导致的染色体区域的失衡。利用具有多个基因座的染色体区域可以得到更高的效率和/或准确度。这种失衡则可以用于潜在癌症患者的诊断或筛查以及癌症患者的预后。可以利用失衡的严重性以及失衡的数量提供癌症的诊断,筛查,预后以及监测。此外,可以在不同的时间点下对患者进行检验以便跟踪一个或多个或众多染色体区域的每一个染
色体区域中的失衡程度以及失衡数量,从而能够进行癌症的筛查和预后、以及监测(例如在治疗后)。
10.根据其中一个实施方案,提供了针对与癌症相关的染色体的缺失或扩增来分析有机体的生物样品的方法。生物样品包括源自正常细胞以及潜在的癌症相关细胞的核酸分子。样品中,至少有一些核酸分子是游离的。在第一染色体区域,针对有机体的正常细胞测定第一和第二单倍型。第一染色体区域包括第一多个杂合基因座。在样品中多个核酸分子的每一个分子在所鉴别的有机体的参照基因组中都具有可识别位置信息和各自的等位基因信息。位置以及所测定的等位基因用于测定源自第一单倍型的第一组核酸分子以及源自第二单倍型的第二组核酸分子。利用计算机系统计算出第一组核酸对应的第一值以及第二组核酸对应的第二值。每个值都定义了各组核酸分子的性质(例如各个组中的分子平均尺寸或数量)。将第一值与第二值比较以便测定第一染色体区域在与癌症相关的任何细胞中是否表现缺失或扩增的分类。
11.根据其中另一个实施方案,提供了分析有机体的生物样品的方法。生物样品包括源自正常细胞以及潜在的癌症相关细胞的核酸分子。样品中,至少一些核酸分子是游离的。有机体的多个非重叠染色体区域被鉴别出来。每个染色体区域包括多个基因座。样品中多个核酸分子的每一个分子都在所鉴别的有机体的参照基因组中具有对应的位置信息。对于每个染色体区域而言,基于所鉴别的位置,各组核酸分子所对应的染色体区域都被识别出来。各组核酸分子包括落在染色体区域的多个基因座的每一个基因座上的至少一个核酸分子。利用计算机系统计算各组中的各值,其中各值定义了各组的核酸分子的性质。将各值与参照值相比,从而测定染色体区域是否存在缺失或扩增。接着定量测定被分类为存染色体组缺失或扩增的染色体区域。
12.根据其中另一个实施方案,提供了使用生物样品测定有机体中染色体畸变进程的方法,其中所述的生物样品包括源自正常细胞以及潜在的癌症相关细胞的核酸分子。在生物样品中,至少一些核酸分子是游离的。针对有机体的参照基因组鉴别一个或多个非重叠的染色体区域。每一个染色体区域都包括多个基因座。分析在不同时间点由有机体取得的样品,以测定病变进程。就样品而言,样品中多个核酸分子的每一个分子都在所鉴别的有机体的参照基因组中具有位置。就各个染色体区域而言,基于所鉴别的位置,各组核酸分子所对应的染色体区域都被鉴别出来。各组核酸分子包括位于染色体区域的多个基因座的每一个基因座上的至少一个核酸分子。计算机系统计算各组核酸分子的各个值。各个值定义了各组核酸分子的性质。将各值与参照值相比,从而测定在第一染色体区域中是否存在缺失或扩增。接着,在多个时间点下利用各染色体区域中的可能缺失或扩增来测定有机体中染色体畸变的进程。
13.本发明的其他实施方案是针对与本文所述的方法相关的系统、便携式设备以及计算机可读介质。
14.参见以下的发明详述以及附图可以更好地理解本发明的特性和优势。
附图说明
15.图1阐述了癌细胞中存在缺失畸变的染色体区域。
16.图2阐述了癌细胞中存在扩增畸变的染色体区域。
17.图3表格300显示了不同类型的癌症、相关的区域及其相应的畸变。
18.图4阐述了根据本发明的实施方案,在血浆中度量在癌细胞内未表现出畸变的染色体区域对应的剂量。
19.图5阐述了根据本发明的实施方案,在血浆中度量在癌细胞内存在缺失的染色体区域510对应的剂量,从而确定对应的缺失区域。
20.图6阐述了根据本发明的实施方案,在血浆中度量在癌细胞内存在扩增的染色体区域610对应的剂量,从而确定扩增对应的区域。
21.图7阐述了根据本发明的实施方案针对定位于染色体1p上的染色体区段,对肝癌(hcc)患者的血浆dna进行的rhdo分析,其中所述的染色体区段显示了在肿瘤组织中存在单等位基因扩增。
22.图8阐述了根据本发明的实施方案,当肿瘤存在染色体缺失时,其染色体区域的两个单倍型各自对应的血浆中的核酸片段长度分布的变化。
23.图9阐明了根据本发明的实施方案,当肿瘤存在染色体扩增时,其染色体区域的两个单倍型各自对应的的血浆中核酸片段长度分布的变化。
24.图10阐明了根据本发明的实施方案,用于分析有机体的生物样品的单倍型来测定染色体区域是否表现出缺失或扩增的方法的流程图。
25.图11说明了根据本发明的实施方案,在癌细胞中存在缺失的区域1110以及子区域1130以及在血浆中对该区域进行的定量测量,从而确定缺失的区域。
26.图12说明了根据本发明的实施方案,如何使用rhdo分析可以绘制畸变的位置。
27.图13说明了根据本发明的实施方案,由另一个方向开始的rhdo的分类。
28.图14为根据本发明的实施方案,使用多个染色体区域来分析有机体的生物样品的方法1400的流程图。
29.图15为根据本发明的实施方案,表格1500阐明了在不同的肿瘤核酸相对百分比浓度下不同数量的畸变片段所需的测序深度。图15提供了针对样品中不同肿瘤核酸相对百分比浓度下所进行的待测分子数量的评估。
30.图16阐述了根据本发明的实施方案,通过相对单倍型剂量(rhdo)分析测量血浆中肿瘤核酸相对百分比浓度的原理。hapi和hapii表示根据本发明的实施方案在非肿瘤组织中的两种单倍型。
31.图17为根据本发明的实施方案,阐述了使用包含核酸分子的生物样品来测定有机体中染色体畸变的进程的方法的流程图。
32.图18a显示出了针对在患有癌症的患者的染色体4的q臂上的染色体区段进行rhdo分析的sprt曲线。圆点表示在各个杂合基因座上游的所有分析位点的累积计数的比值。图18b示出了在针对治疗后患者的染色体4的q臂上的染色体区段进行rhdo分析的sprt曲线。
33.图19显示出了在hcc中发现的常见染色体的畸变。
34.图20a显示出了利用靶向分析(例如,靶向富集捕获测序技术)针对hcc患者和健康对照受试者进行分析,标准化测序读数的比例而得到的结果。图20b显示出了针对3位hcc患者和4位健康对照受试者在靶向富集捕获测序之后得到的血浆中核酸长度分析的结果。
35.图21显示出了根据本发明的实施方案得到的hcc患者的circos图,其描绘了由血浆dna的测序标签计数得到的数据。
36.图22显示出了根据本发明的实施方案,针对未患有hcc的慢性肝炎b病毒(hbv)携带者的血浆样品所进行的测序标签计数分析。
37.图23显示出了根据本发明的实施方案,针对患有第三期鼻咽癌(npc)的患者的血浆样品所进行的测序标签计数分析。
38.图24显示出了根据本发明的实施方案,针对患有第四期npc的患者的血浆样品所进行的测序标签计数分析。
39.图25显示出了根据本发明的实施方案,针对肿瘤组织中存在杂合性缺失(loh)的区域,血浆dna长度累积频率的分布图。
40.图26显示出了δq与loh区域的血浆dna长度之间的关系。根据本发明的实施方案,在长度为130bp时,δq达到0.2。
41.图27显示出了根据本发明的实施方案,在肿瘤组织中存在染色体扩增的区域中的血浆dna长度累积频率分布图。
42.图28显示出了根据本发明的实施方案,针对扩增的区域,δq与血浆dna长度之间的关系。
43.图29显示出了根据本发明的实施方案的系统和方法可用的计算机系统实例900的框图。
44.定义
45.如本文所用,术语“生物样品”是指取自受试者(例如人、患有癌症的人、疑似患有癌症的人、或其他有机体)并包括一种或多种所关注的核酸分子的任何样品。
46.术语“核酸”或“多核苷酸”是指单链或双链形式的脱氧核糖核酸(dna)或核糖核酸(rna),及其聚合物。除非具体限定,否则该术语涵盖了包括天然核苷酸的已知类似物的核酸,该核酸与参照核酸具有相似的结合性质,并且以与天然形成的核苷酸相同的方式代谢。除非另作说明,否则特定的核酸序列还暗示着涵盖了其保守修饰的变体(例如简并密码子取代)、等位基因、直系同源物、单核苷酸多态性(snp)、拷贝数变体、互补序列以及明确地指明的序列。具体而言,简并密码子取代可以通过生成这样的序列来取得,其中在所述的序列中,一个或多个所选的(或所有的)密码子的第三个位置被混合的碱基和/脱氧肌苷残基取代(batzer et al.,nucleic acid res.19:5081(1991);ohtsuka et al.,j.biol.chem.260:2605-2608(1985);和rossolini et al.,mol.cell.probes 8:91-98(1994))。术语核酸涵盖但不局限于:基因、互补dna(cdna)、信使rna(mrna)、小分子非编码rna、微rna(mirna)、piwi-互作rna以及由基因或基因座编码的短发夹rna(shrna)或染色体上其他序列。
47.术语“基因”是指与生产多肽链或转录的rna产物相关的dna的片段。其可以包括编码区域之前及之后的区域(前导区及尾随区),以及单个编码片段(外显子)之间的间隔序列(内含子)。
48.如本文所用,术语“临床相关的核酸序列”或“临床相关的染色体区域”(或者待检验的区域/区段)可以指与其有待检验的潜在的失衡且较大的基因组序列片段或者与其本身的较大的基因组序列相应的多核苷酸序列。其实例包括被删除或扩增的、或者潜在地被删除或扩增的基因组区段(包括简单的复制),或者包括该区段的子区域的较大的区域。在一些实施方案中,多个临床相关的核酸序列、或者临床相关的核酸序列的多个等价的标志
可以提供用于检测区域中的失衡的数据。例如,由染色体上的5个非连续序列得到的数据可以以加和的方式用于测定可能的失衡,从而有效地将所需样品的剂量减少至1/5。
49.如本文所用,术语“参照核酸序列”或“参照染色体区域”是指利用其剂量分布或长度分布与检验区域相比较的核酸序列。参照核酸序列的实例包括不包含缺失或扩增的染色体区域,完整的基因组(例如通过测序标签总计数来归一化),由已知正常的一个或多个样品得到的区域(其与待检验的样品可能是相同的区域),或者特定的单倍型的染色体区域。此类参照核酸序列可以在样品中以内源方式存在,或者在样品处理或分析过程中以外源方式加入。在一些实施方案中,参照染色体区域证明了代表未患有疾病的健康状态的长度分布。在其他实施方案中,证明了参照染色体区域代表未患有疾病的健康状态的定量概况。
50.如本文所用,术语“基于”是指“至少部分基于”并且指在另一个值的测定中所使用的一个值(或者结果),例如在一种方法的输入与该方法的输出的关系中所形成的。如本文所用,术语“推导(derive)”还指一种方法的输入与该方法的输出的关系,例如在推导为公式的计算时所形成的。
51.如本文所用,术语“参数”是指表征了定量数据组和/或定量数据组之间的数量关系的数值。例如,第一核酸序列的第一量与第二核酸序列的第二量的比值(或比值的函数)为参数。
52.如本文所用,术语“基因座”为在基因组中可以具有变化的任何长度的核苷酸(或碱基对)的位置或地址。
53.如本文所用,术语“序列失衡”或“畸变”是指临床相关的染色体区域的量与参照量的所定义的至少某一个阈值存在的任何显著的偏离。序列失衡可以包括染色体剂量失衡、等位基因失衡、突变剂量失衡、拷贝数失衡、单倍型剂量失衡以及其他相似的失衡。例如,等位基因失衡的形成可以是由于肿瘤具有一个所删除基因的等位基因或一个所扩增基因的等位基因,或者在其基因组中两个等位基因的差异扩增,由此在样品的特定基因座处形成了失衡。又例如,患者在肿瘤抑制基因中可以具有遗传突变。则该患者可以继续发展成肿瘤,其中肿瘤抑制基因的非突变等位基因被删除。因此,在肿瘤中,存在突变剂量失衡。当肿瘤将其dna释放到患者的血浆中时,肿瘤dna将与患者的正常体细胞结构dna在血浆中混合。通过使用本文所述的方法,可以检测血浆中该dna混合体存在的突变剂量失衡。
54.如本文所用,术语“单倍型”是指在多个基因座处等位基因的组合,其中所述的多个等位基因在相同的染色体或染色体区域一起传递给子代。单倍型可以指少至一对的基因座或染色体区域,或完整的染色体。术语“等位基因”是指在相同染色体上的相同物理位置处备选的dna序列,其可能得到相同或不同的表型特征。在任何特定的二倍体有机体中,其各染色体具有两个拷贝(除了男性受试者中的性染色体),各基因的基因型包括出现在该基因座处的成对的等位基因,其相同则为纯合体,而其不同则为杂合体。有机体群体或物种在不同的个体中在各个基因座处通常包括多种等位基因。其中在群体中发现多于一种等位基因的基因座被称为多态性位点。基因座处等位基因的变化程度可以用等位基因的数量(即,多态性的程度)或者群体中杂合体的比例(即,杂合性的比例)来度量。如本文所用,术语“多态性”是指人类基因中任何个体间的变化,而与其频率无关。此类变化的实例包括但不局限于单核苷酸多态性、简单串联重复多态性、插入-缺失的多态性、突变(其可能是疾病的原因)及拷贝数的变化。
55.术语“测序标签”是指由核酸分子的全部或部分(例如dna片段)测定的序列。通常,仅片段的一个末端被测序,例如大约30bp。然后,将测序标签比对到参照基因组。另一种方式,可以测序片段的两个末端,从而生成两个测序标签,可以提供更高的比对准确度,并且还可以提供片段长度信息。
56.术语“通用测序”是指将已知的接头序列连接到片段的末端而用于测序的引物可以互补配对到接头序列上进行测序。因此,任何片段可以使用相同的引物来测序,因此测序可以是随机的。
57.术语“尺寸分布”是指表示与特定组(例如由特定单倍型或特定染色体区域得到的片段)相应的分子的长度、质量、重量或尺寸的其他量度的任意一个值或一组值。多种实施方案可以使用多种尺寸分布。在一些实施方案中,尺寸分布涉及一个染色体相关的片段相对于其他染色体相关的片段的长度相对秩序(例如平均值、中值或几何平均值)。在其他实施方案中,尺寸分布可以涉及染色体片段的实际尺寸的统计值。在一个实施方式中,统计值可以包括染色体片段的任何平均值、几何平均值或中值。在另一个实施方式中,统计值可以包括某个阈值以下的片段的总长度,其可以除以所有片段或至少某个较大截断值以下的片段的总长度。
58.如本文所用,术语“分类”是指与样品的特定性质相关的任何数量或其他特征。例如,“+”符号(或词语“阳性”)可以表示样品被分类为存在缺失或扩增的染色体畸变。分类可以为二分类(例如正的或负的),或者具有更高水平的分类(例如1至10或0至1的级别)。术语“截断”和“阈值”是指在操作中使用的预定值。例如,截断尺寸可以指高出该值所对应的片段将会被排除。阈值可以为这样的值,高于或低于该值,特定的分类可以应用。这些术语的任意一个可以在这些内容的任意内容中使用。
59.术语“癌症的水平”可以指癌症是否存在、癌症的阶段、肿瘤的尺寸、涉及染色体区域的缺失或扩增的程度(例如双重复制扩增或三重复制扩增)、和/或癌症严重性的其他量度。癌症的水平可以为数量或其他特征。该水平可以为零。此外,癌症的水平还包括与缺失或扩增相关的恶化前或癌症前的状态。
60.发明详述
61.癌症组织(肿瘤)可以具有畸变,例如染色体区域的缺失或扩增。肿瘤可以将dna片段释放到机体的流体组织中,如外周血。多个实施方案可以通过相对于染色体区域中dna的正常值(期望值)来分析dna片段,从而鉴别畸变,由此鉴别肿瘤。
62.缺失或扩增的确切尺寸以及位置可以改变。或许特定的染色体区域存在对癌症或某特定癌症而言常见的已知畸变(由此可以诊断特定的癌症)。当特定的区域是未知时,可以使用用于分析整个基因组或大部分的基因组的系统性方法来检测畸变区域,该区域可能分散在整个基因组中并且其尺寸(例如所缺失或扩增的碱基的数量)是变化的。可以在不同的时间点下跟踪染色体区域,从而检测一个畸变的严重程度的变化情况或检测一些畸变区域的变化情况。这种跟踪可以提供用于癌症筛查、预后诊断及监测肿瘤的重要信息(例如在治疗后或者用于检测复发或肿瘤进程)。
63.该发明详述首先以癌症中染色体畸变的实例开始。接着,讨论通过检测和分析生物样品中游离dna来检测染色体畸变的方式的实例。一旦建立检测一个染色体区域中畸变的方法,则进一步描述如何将许多染色体区域中的畸变检测以系统性方式用于癌症患者的
筛查(诊断)和预后所对应的方法。此外,该发明详述还描写了在多个时间点下检测一个或多个区域来跟踪由检验染色体畸变获得的癌症相关的数量指标所对应的方法,从而提供对患者的筛查、预后和监测。接着讨论实例。
64.i.癌症中染色体畸变的实例
65.染色体畸变在癌细胞中是普遍存在的。此外,特定的癌症具有特定的染色体畸变的特征模式。例如,染色体臂1p,1q,7q,15q,16p,17q和20q中对应的dna数量的增加以及3p,4q,9p和11q处对应的dna的数量缺失通常在肝细胞癌(hcc)中检测到。之前的研究已经证明此类遗传畸变还可能在癌症患者的外周血dna中检测到。例如,在患有肺癌及头颈癌的患者的外周血dna分子中,已有研究指出在特定的基因座处检测出杂合性缺失(loh)(chen xq,et al.nat med 1996;2:1033-5;nawroz h,et al.nat med 1996;2:1035-7)。在血浆或血清中检测的遗传变异与在肿瘤组织中发现的遗传变异相一致。但是,由于肿瘤衍生的dna仅构成了外周血总的游离dna的较小的一部分,则由肿瘤细胞的loh所导致的等位基因失衡通常是微弱的。许多研究员已经开发了数字聚合酶链式反应(pcr)技术(vogelstein b,kinzler kw.proc natl acad sci u s a.1999;96:9236-41;zhou w,et al.nat biotechnol 2001;19:78-81;zhou w,et al.lancet.2002;359:219-25),以便用于在外周血dna分子中进行准确地定量基因座上的不同等位基因(chang hw,et al.j natl cancer inst.2002;94:1697-703)。与实时pcr或用于检测肿瘤dna中特定基因座处由loh所导致的微弱的等位基因失衡的其他dna定量方法相比,数字pcr要更加敏感。但是,数据pcr在鉴别特定基因座处的极微弱的等位基因失衡中仍具有困难,因此本文所述的实施方案以综合的方式分析了染色体的不同的区域。
66.此外,本文所述的技术可以应用于检测恶化前或癌症前的状态。此类状态的实例包括肝硬化及宫颈上皮内瘤样病变。肝硬化的状态为肝癌恶化前的状态,而宫颈上皮内瘤样病变的状态为宫颈癌恶化前的状态。据报导,此类恶化前的状态在其演变成为恶性肿瘤过程中已经具有多种分子改变。例如,loh在染色体臂1p,4q,13q,18q处的存在、以及在多于3个基因座的同时缺失与患有肝硬化的患者中hcc发生风险的增高有关(roncalli m et al.hepatology 2000;31:846-50)。此外,此类恶化前的病变还会将dna释放到外周血循环系统中,但是可能为较低的浓度。所述的技术可以通过分析血浆中的dna片段并测量血浆中恶化前游离dna的浓度(包括相对百分比浓度)来检测缺失或扩增。此类畸变可以容易地检测(例如测序深度或者所检测的此类变化的数量),并且浓度能预测恶化成全面爆发的癌症状态的可能性或速度。
67.a.染色体区域的缺失
68.图1阐述了癌细胞中存在缺失畸变的染色体区域。正常细胞显示成具有两种单倍型hap i和hap ii。如图1所示,在多个杂合基因座110(也称为单核苷酸多态性snp)的每个基因座中,hap i和hap ii都具有序列信息。在与癌症相关的细胞中,hap ii具有缺失的染色体区域120。例如,与癌症相关的细胞可以得自肿瘤(例如恶性肿瘤),得自肿瘤的转移病灶(例如在局部淋巴结中或者在远端器官中),或者得自癌症前或恶化前的病变,例如如上文所述的那些病变。
69.在癌细胞的染色体区域120中,其中两个同源单倍型之一被删除,由于在被删除的同源染色体上其他相应的等位基因的缺失,则所有的杂合snps 110均表现为纯合的。因此,
这种类型的染色体畸变被称为杂合性缺失(loh)。在区域120中,这些snp的非删除的等位基因表示可以在正常组织中找到的两个单倍型中的一者。在图1所示的实例中,可以通过肿瘤组织的基因型来测定loh区域120处的单倍型i(hap i)。可以通过将正常组织的基因型与癌症组织的表面上的基因型相比较来测定其他的单倍型(hap ii)。可以通过连接所有的缺失的等位基因来构建hap ii。即只在正常细胞的区域120处存在的等位基因(但其在癌细胞的区域120处处于缺失状态)被测定为相同的单倍型,即hap i。通过该分析,可以测定肿瘤组织中表现出loh的所有染色体区域所对应的患者的单倍型(例如肝细胞癌hcc患者)。此类方法只对癌细胞分析是有用的,并且仅对用于测定区域120中的单倍型有效,但是其很好地阐明了染色体中缺失区域。
70.b.染色体区域的扩增
71.图2阐述了癌细胞表现出扩增畸变的染色体区域。正常细胞显示出具有两种单倍型hap i和hap ii。如图2所示,在多个杂合基因座210的每个基因座中,hap i和hap ii都具有序列信息。在肿瘤细胞中,hap ii具有扩增2倍的染色体区域220(复制的)。
72.类似地,对于肿瘤组织中具有单等位基因扩增的区域,可以通过诸如芯片分析之类方法来检测snps 210处扩增的等位基因。可以通过将染色体区域220中所有扩增的等位基因连接一起来测定两个单倍型中的一种单倍型(如图2所示实例中的hap ii)。可以通过将比较基因座处各等位基因的数量来测定特定基因座处的扩增的等位基因。接着,可以通过将非扩增的等位基因连接一起来测定其他的单倍型(hap i)。此类方法只有对癌细胞分析是有用的,并且仅对于测定区域220中的单倍型有效,但是其很好地阐明了染色体中的扩增区域。
73.扩增可以得自多于2个染色体的区域,或者得自于一个染色体中的某个基因的重复。一个区域可以被串联复制,或者一个区域可以是包括一个或多个拷贝的区域的微小染色体。此外,扩增还可以得自被复制了的一个染色体的基因,并且该复制产物被插入到不同的染色体或者同一染色体的不同区域中。此类插入为一种类型的扩增。
74.ii.染色体区域的选择
75.当癌组织至少贡献了一部分游离dna(以及潜在的胞内dna)时,可以在诸如血浆和血清之类的样品中检测癌组织的基因组畸变。检测所述的畸变所存在的难题在于肿瘤或癌症可能是相当小的,因此由癌细胞提供的dna相对较微弱。因此,具有畸变的游离dna的量是相对较少的,由此使得检测变得极其困难。在待检测畸变的基因组中的单一基因座处,可能不具有足够的dna。本文所述的方法可以通过分析包括多个基因座的染色体区域(单倍型)处的dna,由此基于单倍型将各个基因座处的微小的改变聚集成可觉察的差异来克服所述的困难。由此,分析一个区域中的多个基因座可以提供更高的准确度和精确度,并可以减少假阳性和假阴性。
76.此外,畸变的区域可能是相当微小的,由此难以鉴别畸变。如果仅使用一个基因座或特定的基因座,则不在这些基因座上的畸变将被遗漏。如本文所述,一些方法可以用于研究整个区域,从而发现存在于这些区域的子区域中的畸变。当所分析的区域覆盖整个基因组时,由此可以分析整个基因组从而发现不同长度及不同位置的畸变,如下文中更加详细描述的那样。
77.为了说明这些观点,如上文所示,区域可以具有畸变。但是,必须选择用于分析的
区域。区域的长度和位置可以改变结果,因此影响分析。例如,如果分析图1所示的第一区域,则没有畸变被检测到。如果分析第二区域,例如使用本文所述的方法,则可以检测到畸变。如果分析包括第一区域和第二区域的较大的区域,人们会遇到这样的难题,即,仅仅较大的区域中的一部分具有畸变,这可能更难以鉴别任何畸变,并且人们会遇到鉴别畸变的确切位置和长度的问题。本发明的多种实施方案可以解决一些和/或所有的这些难题。用于选择区域的描述同样适用于使用相同染色体区域的单倍型或者使用两个不同染色体区域的单倍型方法。
78.a.选择特定的染色体区域
79.在一个实施方案中,可以根据癌症或患者的了解来选择特定的区域。例如,已知该区域在许多癌症或特定的癌症中普遍存在的畸变。相对应的区域的确切长度和位置的获得可以基于参考一些与癌症的类型或者患者所具有的特定的风险因子相关的知名文献。此外,可以获得并分析患者的肿瘤组织,从而鉴别畸变的区域,如上文所述。目前,此类技术须要获得癌细胞(这对于刚刚被诊断的患者可能是不实际的),但是此类技术可以用于鉴别在相同的患者中而在不同的时间点下进行监测的区域(例如在手术除去癌组织后,或者在化疗或免疫治疗或靶向治疗后,或者用于检测肿瘤复发或进程)。
80.人们能够鉴别多于一个的特定区域。可以独立地使用每一个此类区域进行分析,或者可以共同分析不同的区域。此外,可以细分这些区域,从而在定位畸变中提供更高准确度。
81.图3显示出了在表格300中显示不同类型的癌症、相关的区域及其相应的畸变。列310列出了不同的癌症类型。本文所述的实施方案可以用于与畸变有关的任何类型的癌症,因此,该列表仅为实例。列320显示出了多个区域(例如大的区域,例如7p或者更特异的区域例如17q25),其中增加(扩增)与同一行的特定癌症有关。列330示出了其中可以发现缺失(删除)的区域。列340列出了参考文献,这些文献讨论了这些区域与特定癌症的相关性。
82.根据本文所述的方法,对于具有潜在的染色体畸变的这些区域而言,可以将其当作应用于畸变分析的候选染色体区域。在癌症中发生改变的其他基因组区域的实例可以在cancer genome anatomy project(cgap.nci.nih.gov/chromosomes/recurrentaberrataions)以及atlas of genetics and cytogenetics in oncology and haematology(atlasgeneticsoncology.org tumors/tumorliste.html)的数据库中找到。
83.正如人们可以看见的那样,所鉴别的区域可以相当大,而其他区域可以是更特异性的。畸变不一定包括表中所鉴别的整个区域。因此,对于特定患者而言,该类型畸变的此类线索不能被确切地精确定位,但是更多地可以用作用于分析的大区域的粗略指导。此类大区域可以包括许多子区域(它们可以是相等的尺寸)。其可以用于单独分析以及共同分析(其详细情况在本文中描述)。因此,基于待检验的癌症的具体情况,多个实施方案可以将选择大区域的多个方面结合起来,但是还可以使用如下文所述的更通用的技术。
84.b.选择任意的染色体区域
85.在另一个实施方案中,任意地选择待分析的染色体区域。例如,基因组可以分成长度为1兆碱基(mb)的区域,或者其他预定片段的长度,例如500kb或2mb。如果区域为1mb,因为在单倍体人类基因组中存在大约30亿个碱基,则在人类基因组中存在大约3000个区域。正如下文更加详细的讨论,这些区域的每一个都可以被分析。
res 19:3561-3567)、克隆及限制性酶消化(smirnova as et al.immunogenetics 2007;59:93-8)等的单倍型分析技术。其他方法是基于群体中单倍型区块的分布及连锁不平衡结构,其允许受试者的单倍型可以由统计学评估推导而得到(clark ag.mol biol evol 1990;7:111-22;10:13-9;salem rm et al.hum genomics 2005;2:39-66)。
94.测定loh的区域的单倍型的另一个方法是通过对受试者的正常组织和肿瘤组织(如果肿瘤组织是可获得的)进行基因型分型。在loh存在下,肿瘤细胞的相对百分比浓度极高的肿瘤组织在存在loh的区域内的所有snp基因座均会显示出表观纯合性。这些snp基因座的基因型包括一种单倍型(图1所示的loh区域的hap i)。另一方面,在正常组织中,其loh的区域内的snp基因座会显示出杂合性。存在于正常组织而不存在于肿瘤组织内的等位基因包括其他单倍型(图1所示的loh区域的hap ii)。
95.b.相对单倍型剂量(rhdo)分析
96.如上文提及的那样,具有染色体区域的单倍型之一的扩增或缺失的染色体畸变会导致肿瘤组织中对应染色体区域中的两种单倍型的剂量失衡。在具有肿瘤生长的人的血浆中,一部分的外周血dna是源自于肿瘤细胞的。由于癌症患者的血浆中存在肿瘤衍生的dna,所以此类失衡还会存在于他们的血浆中。两种单倍型的剂量的失衡可以通过计数源自各种单倍型的分子的数量来检测。
97.就在肿瘤组织中观察到loh的染色体区域(例如图1所示的区域120)而言,由于缺乏由肿瘤组织的hap ii的贡献,所以当与hap ii相比时,在外周血dna分子(片段)中,hap i是相对呈现过量。就在肿瘤组织中观察到拷贝数扩增的染色体区域而言,由于肿瘤组织释放出额外hap ii的剂量,所以就受到hap ii的单等位基因扩增的影响的区域而言,当hapii与hap i相比较时,hap ii是相对呈现过量。为了测定呈现过量或呈现不足的情况,可以通过多种方法进行测定样品中的某些得自hap i或hap ii的dna片段,例如基于利用通用测序及比对分析,或者使用数字pcr及序列特异性的探针等若干方法。
98.在对由癌症患者的血浆(或其他生物样品)得到的多个dna片段进行测序从而生成经序列标签之后,可以识别并计数与两种单倍型上的等位基因相应的测序标签。然后,比较与两种单倍型的每一种相应的测序标签的数量从而测定两种单倍型是否相等地存在于血浆中。在一个实施方案中,可以使用序贯概率比检验(sprt)来测定血浆中的两种单倍型的呈现是否存在显著性差异。统计学显著性差异表明在所分析的染色体区域中存在染色体畸变。此外,血浆中两种单倍型在定量上的差异可以用于估计血浆中肿瘤衍生的dna的相对百分比浓度,如下文所述。
99.本技术所述的用于测定dna片段的特性(例如其在人类基因组中的位置)的诊断方法不局限于根据本发明的实施方案使用大规模平行测序作为检测平台。此外,这些诊断方法还可以用于例如但不局限于微流体数字pcr系统(例如流体数字阵列系统)、微滴式数字pcr系统(例如raindance and quantalife)、beam-ing系统(即基于珠、乳液pcr、扩增和磁学的系统)(diehl et al.procnatlacadsci usa 2005;102:16368-16373)、实时pcr、基于质谱的系统(例如sequenommassarray系统)以及多重连接依赖的探针扩增(mlpa)分析。
100.正常区域
101.图4显示出了根据本发明的实施方案,在未表现出畸变的癌细胞内的染色体区域以及在血浆中进行的测量。可以通过任何方法选择染色体区域410,例如基于待检验的特异
癌症的方法或者基于通用的筛查的方法(即其使用了覆盖大部分基因组的预定片段对应的方法)。为了区分两种单倍型,首先测定两种单倍型。图4显示出了就染色体区域410而言,正常细胞的两种单倍型(hap i和hap ii)。单倍型包括第一多个基因座420。该第一多个基因座420跨越待分析的染色体区域410。如所示,这些基因座在正常细胞中是杂合的。癌细胞的两种单倍型也被显示出来。在癌细胞中,没有区域被删除或扩增。
102.此外,图4还显示出了就各个基因座420而言,在各个单倍型上等位基因计数的数量。此外,还提供了染色体区域410的某些子区域的累积总数。等位基因计数的数量与dna片段的数量(其与在各个特定基因座处的特定单倍型相应)相应。例如,包含第一基因座421并具有等位基因a的dna片段取得hap i的计数。而具有等位基因t的dna片段取得对hap ii的计数。可以以多种方式测定片段比对到何处(即,其是否包括特定的基因座)以及其包括何种等位基因,如本文所提及。在两种单倍型上的计数比可用于测定是否存在统计学显著性差异。该计数比在本文中也称为比值比。此外,还可以使用两个值之间的差异;可以将差异对片段的总数归一化。比值和差异(及其函数)为参数的实例,将该参数与阈值相比较从而测定是否存在畸变的分类。
103.rhdo分析可以利用相同单倍型上的所有等位基因(例如累积计数)来测定在血浆中是否存在两种单倍型的任何失衡,例如可以在母亲的血浆中实施,如在lo的专利申请12/940,992和12/940,993中所述,参见上文。该方法可以显著地增加用于测定是否存在任何失衡的dna分子的数量,可以用于从非癌症的随机波动中区分出癌症导致的失衡(由于在缺乏癌症或恶化前状态的等位基因计数是随机分布而癌症导致的失衡是既定的)并因此得到较好的统计功效。与单独分析多个snp基因座相比,rhdo方法可以利用两条染色体上等位基因的相对位置(单倍型信息),从而可以一起分析位于同一染色体上的等位基因。在缺乏单倍型信息的情况下,不同snp基因座的等位基因计数不能加在一起地以这种统计学方式测定单倍型在血浆中是否是呈现过量或呈现不足。等位基因计数的定量可以通过但不局限于大规模平行测序(例如illumina变合成边测序测序系统,通过life technologies的连接边测序(solid)的系统,通过ion torrent and life technologies的ion torrent测序系统,纳米孔测序(nanoporetech.com)技术,454测序技术(roche),数字pcr(例如通过微流体数字pcr(例如流体(fluidigm.com))),beaming(珠、乳液pcr、扩增、磁性(inostics.com)),或微滴式数字pcr(例如通过quantalife(quantalife.com)and raindance(raindancetechnologies.com))以及实时pcr。在所述技术的其他实施方式中,可以使用高通量靶标捕获测序(其利用了液相捕获(例如利用了agilent sureselect system,the illumina truseq custom enrichment kit(illumina.com/applications/sequencing/targeted_resequencing.ilmn),或通过mygenostics gencap custom enrichment system(mygenostics.com/)))或基于阵列的捕获(例如使用roche nimblegen系统)。
104.在图4所示的实例中,观察到前两个snp基因座(就第一snp而言为24与26,对第二snp而言为18与20)的轻微的等位基因失衡。但是,等位基因计数的数量在统计学上不足以测定是否存在真实的等位基因失衡。因此,将同一单倍型上的等位基因的计数加在一起,直到两种单倍型的等位基因的累积计数足以得到统计学上的结论:在染色体区域410上,两种单倍型之间不存在等位基因失衡(就该实例而言,为第五snp)。在达到统计学意义的分类以后,重新设定累积计数(就该实例而言,在第六snp处)。然后测定累积计数,直到两种单倍型
的累积等位基因的计数再次足以得到统计学上的结论:就区域410的特定子区域而言,两种单倍型之间不存在等位基因失衡。总的累积计数还可以用于整个区域,但是之前的方法可以允许不同的子区域被检验,与整个区域410不同,上述检验在测定畸变的位置中提供了更高的精确度(即子区域)。用于测定是否存在真实的等位基因失衡的统计学检验的实例包括但不局限于序贯概率比检验(zhou w,et al.nat biotechnol 2001;19:78-81;zhou w,et al.lancet.2002;359:219-25)、t检验和chi-square检验。
105.检测删除
106.图5阐述了根据本发明的实施方案,在癌细胞内染色体区域510的缺失以及在血浆中进行的测量,从而测定存在缺失的区域。图5示出了正常细胞的染色体区域510的两种单倍型(hap i和hap ii)。单倍型包括第一多个杂合基因座520,该基因座覆盖了待分析的染色体区域510。此外,还示出了癌细胞的两种单倍型。在癌细胞中,就hap ii而言,删除了区域510。如图4类似,图5还示出了各个基因座520的等位基因计数的数量。此外,就染色体区域510内的某些子区域而言,还记录了累积的总量。
107.由于肿瘤组织通常包括肿瘤细胞和非肿瘤细胞的混合物,所以可以通过区域510内的基因座处,两种等位基因的量的比例的相对偏移来证实loh。在这种情况下,可以通过基因座520的组合来测定区域510中被删除的单倍型hap ii,其与正常组织上的相应基因座相比较,所述的组合显示dna片段的量相对减少。单倍型片段显现频率较高的为hap i,其被保留在肿瘤细胞中。在某些实施方案中,理想的是实施这样的过程:该过程会富集肿瘤样品中肿瘤细胞的比例,从而允许缺失及保留的单倍型更容易地测定。此类过程的一个实例为显微切割(以人工方式或通过激光捕获技术)。
108.理论上,就肿瘤组织中表现出loh的染色体区域而言,hap i上的每一个等位基因在外周血dna中是呈现相对过量形式,并且等位基因失衡的程度取决于血浆中肿瘤dna的相对百分比浓度。但是,同时外周血dna的样品中两种等位基因的相对量服从poisson分布。可以利用统计学分析来测定所观察的等位基因失衡是否是由于癌组织中loh的真实存在或由于偶然波动。检测癌症中与loh相关的等位基因失衡的能力取决于待分析的外周血dna分子的数量以及肿瘤dna的相对百分比浓度。肿瘤dna相对百分比浓度越高,以及用于分析的分子数目越多,得到用于检测等位基因失衡的敏感性和特异性将越高。
109.在图5所示的实例中,观察到前两种snp基因座(就第一snp而言为24与22,对第二snp而言为18与15)的轻微的等位基因失衡。但是,等位基因计数的数量在统计学上不足以测定是否存在真实的等位基因失衡。因此,将同一单倍型上的等位基因的计数加在一起,直到两种单倍型的累积等位基因计数足以得到统计学上的结论:在区域510上,两种单倍型之间存在等位基因失衡(就该实例而言,为第五snp)。在一些实施方案中,仅知道失衡,并且未测定特定的类型(缺失或扩增)。然后,测定累积计数,直到两种单倍型的累积等位基因的计数再次足以得到统计学上的结论:就区域510的特定子区域而言,两种单倍型之间存在等位基因失衡。总的累积计数还可以用于整个区域,其可以以本文所述的任何方法实施。
110.检测染色体区域的扩增
111.图6阐述了根据本发明的实施方案,在癌细胞内染色体区域610的扩增以及在血浆中进行的测量,从而测定扩增的区域。除了loh以外,在癌组织中还频繁地观察到染色体区域的扩增。在图6所示的实例中,染色体区域610中的hap ii在癌细胞中扩增至3个拷贝。如
图所示,区域610仅包括6个杂合的基因座,这与之前的附图中所示的更长的区域不同。在6个基因座中,扩增被鉴定为具有统计学上显著性,其中呈现过量被测定为具有统计学上显著性。在一些实施方案中,仅知失衡,并且未测定特定的类型(缺失或扩增)。在其他实施方案中,可以获得癌细胞并进行分析。此类分析可以提供关于失衡是否由于缺失(就删除的区域而言,癌细胞是纯合的)或扩增(就扩增的区域而言,癌细胞是杂合的)的信息。在其他实施方式中,可以使用部分iv中所述的方法来测定是否存在删除或扩增,从而分析完整的区域(即并非分析各个单倍型)。如果区域是过多呈现的,则畸变为扩增;如果区域是呈现不足的,则畸变是删除的。此外,还分析区域620,并且累积计数证明不存在失衡。
112.用于血浆rhdo分析的sprt分析
113.就具有杂合基因座的任何染色体区域而言,可以使用rhdo分析来测定血浆中是否存在两种单倍型之间的任何剂量失衡。在这些区域中,血浆中单倍型剂量失衡的存在表明血浆样品中存在肿瘤衍生的dna。在一个实施方案中,sprt分析可以用于测定hap i及hap ii的测序读数的数量差异是否具有统计学上的显著性。在sprt分析的实例中,我们首先测定每一种单倍型对应的测序读数的数量。然后,我们测定参数(例如百分比浓度),该参数表示由潜在的呈现相对过量的单倍型所贡献的测序读数的比例(例如一种单倍型的读数的数量除以其他单倍型的读数的数量的比例)。在loh的方案中,潜在的呈现过量的单倍型是非删除的单倍型,而在染色体区域的单等位基因扩增的方案中,潜在的过多呈现的单倍型是扩增的单倍型。接着,将该比例与两个阈值(上限阈值和下限阈值)比较,这些阈值是基于零假设(即不存在单倍型剂量失衡)和备择假设(即存在单倍型剂量失衡)而构建的。如果该比例大于上限阈值,则其表示血浆中的两种单倍型间存在统计学上显著性的失衡。如果该比例低于下限阈值,则其表示两种单倍型间不存在统计学上显著性的失衡。如果该比例介于上限阈值和下限阈值之间,则其表示不具有足够的统计学证据来得出结论。就待分析的区域而言,可以将杂合基因座的数量的连续累加直到可以成功进行sprt分类。
114.用于计算sprt的上界限和下界限的数学公式为:上限阈值=[(ln8)/n
–
lnδ]/lnγ;下限阈值=[(ln 1/8)/n
–
lnδ]/lnγ,其中δ=(1
–
θ1)/(1
–
θ2)而θ1为在血浆中存在等位基因失衡时,由潜在的呈现过量的单倍型对应的测序标签的期望比值,θ2为在不存在等位基因失衡时,两种单倍型的任意一者的期望比值,即0.5;n为hap i和hap ii的测序标签的总数;ln为表示自然对数的数学符号,即loge。θ1取决于血浆中肿瘤dna的相对百分比浓度(f)。
[0115]
在loh的方案中,θ1=1/(2-f)。在单一等位基因扩增的方案中,θ1=(1+zf)/(2+zf),其中z表示在肿瘤中扩增的染色体区域对应的额外增加的拷贝数。例如,如果一个染色体被复制,则为特定染色体的一个额外增加的拷贝。那么z等于1。
[0116]
图7显示出了根据本发明的实施方案,针对位于染色体1p上的染色体区段,对hcc患者的血浆dna进行的rhdo分析,其中所述的染色体区段在肿瘤组织中具有单等位基因的扩增。绿色三角形表示患者的数据。测序读数的总数随着待分析的snp的数量的增多而增多。得自于肿瘤中染色体扩增畸变所对应的单倍型的总测序读数的比列随着所分析的总测序读数的增大而变化,并最终达到高于上限阈值的值。这表明存在显著性单倍型剂量失衡,并因此佐证了血浆中存在这种癌症相关的染色体畸变。
[0117]
对hcc患者的所有染色体区域利用基于sprt的rhdo分析,其中所述的区域在肿瘤组织中显示出扩增和缺失。对于已知具有loh的922个染色体区段以及已知具有扩增的105染色体区段分别进行rhdo分析,结果如下。对于loh,使用sprt对922个染色体区段进行分类,而其中的921个区段正确地被鉴别为在血浆中具有单倍型剂量失衡,从而提供99.99%的准确度。对于单等位基因扩增,使用sprt对105个区段进行分类,而其中的105区段正确地被鉴别为在血浆中具有单倍型剂量失衡,从而提供100%的准确度。
[0118]
c.相对的单倍型尺寸的分析
[0119]
利用单倍型的各自对应片段的长度可以作为利用两种单倍型所对应的片段计数剂量的备选方法。例如,对于特定的染色体区域,由一个单倍型得到的dna片段的尺寸可以与其他单倍型的dna片段的尺寸相比较。人们可以分析在区域的第一单倍型的杂合基因座处与任一等位基因相应的dna片段的尺寸分布,并将其与第二单倍型的杂合基因座处与任一等位基因相应dna片段的尺寸分布相比较。在尺寸分布中具有统计学显著性差异可以利用与剂量计数的相同方式来鉴别畸变。
[0120]
据报导,总的血浆dna的尺寸分布(即肿瘤与非肿瘤的)在癌症患者中升高(wang bg,et al.cancer res.2003;63:3966-8)。但是,如果人们专门地研究肿瘤衍生的dna(非总的dna(即即肿瘤与非肿瘤的)),则观察到肿瘤衍生的dna分子的长度分布小于由非肿瘤细胞衍生的分子的长度分布(diehl et al.proc natl acad sci u s a.2005;102:16368-73)。因此,外周血dna的尺寸分布可以用于测定癌症相关的染色体畸变是否存在。尺寸分析的原理示于图8中。
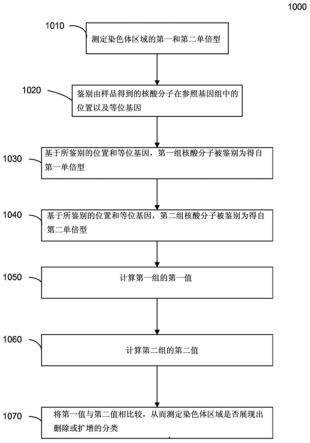
[0121]
图8示出了根据本发明的实施方案,当包括缺失畸变的肿瘤存在时,就染色体区域的两个单倍型而言片段尺寸分布的变化。如图8所示,在肿瘤组织中,t等位基因被删除。结果,肿瘤组织仅将a等位基因的短分子释放到血浆中。肿瘤衍生的短dna分子会导致血浆中a等位基因对应的长度分布整体缩短,因此使得血浆中,a等位基因的长度分布比t等位基因的长度分布更短。如之前的部分所讨论的那样,位于相同单倍型上的所有等位基因可以一起分析。换言之,携带位于一个单倍型上的等位基因的dna分子的尺寸分布可以与携带位于其他单倍型上的等位基因的dna分子的尺寸分布相比较。肿瘤组织中缺失的单倍型在血浆中显示出更长的尺寸分布。
[0122]
此外,尺寸分析还可以适用于检测与癌症相关的染色体区域的扩增。图9示出了根据本发明的实施方案,当包括扩增畸变的肿瘤存在时,就染色体区域的两个单倍型而言片段尺寸分布的变化。在图9所示的实例中,携带等位基因t的染色体区域在肿瘤被复制。结果,携带t等位基因的较短dna分子被释放到血浆中的量有所增加,因此使得t等位基因对应片段的尺寸分布比a等位基因对应片段的尺寸分布从整体上来讲显得更短。与上述类似,位于相同单倍型上的所有等位基因可以一起分析。换言之,在肿瘤组织中扩增的单倍型的尺寸分布比在肿瘤中未扩增的单倍型的尺寸分布显得更短。
[0123]
外周血dna的尺寸分布缩短的检测
[0124]
由两种单倍型(即hap i和hap ii)得到的dna片段的尺寸可以通过但不局限于双末端大规模平行测序(paired-end massively parallel sequencing)来测定。在对dna片段的末端进行测序之后,可以将测序读数(标签)与人类参照基因组比对。可以由各个末端处最外侧核苷酸的坐标来推导出测序dna分子的尺寸。分子的测序标签可以用于测定测序
dna片段是否从hap i或hap ii得到。例如,测序标签之一可以包括待分析的染色体区域中的杂合基因座。
[0125]
因此,就各个测序分子而言,我们可以测定其长度以及其是否由hap i或hap ii得到。基于比对到各自单倍型的片段尺寸,计算机系统可以计算hap i和hap ii的尺寸分布(例如平均片段尺寸)。可以使用合适的统计学分析来比较由hap i和hap ii得到的dna片段的尺寸分布,从而测定尺寸分布是否有足够的不同,从而可以鉴别畸变存在与否。除了双末端大规模平行测序,其他方法可以用于测定dna片段的尺寸,包括但不局限于测序整个dna片段、质谱以及用于观察和将所观察得到的dna分子长度与标准相比较的光学方法。
[0126]
接着,我们引入了用于测定与肿瘤的基因畸变相关的外周血dna的缩短的两个实例方法。这两种方法的目的在于对两个群体的dna片段提供尺寸分布的差异的定量测量。两个群体的dna片段是指与hap i和hap ii相应于的dna分子。
[0127]
短的dna片段的比例的差异
[0128]
在一个实施方式中,使用短的dna片段的比例。人们设定了长度阈值(w),从而定义短的dna分子。可以改变并选择不同的长度阈值,从而拟合不同的诊断目的。计算机系统可以测定与长度阈值相等或较短的分子的数量。然后,可以通过将短的dna的数量除以dna片段的总数量来计算短的dna片段的比例(q)。q值会受到dna分子的群体的长度分布的影响。较短的整体尺寸分布表明较高比例的dna分子为短片段,因此得到较高的q值。
[0129]
接着,可以使用hap i和hap ii之间短dna片段的对应比例的差异。就hap i和hap ii而言,可以由短片段比例的差异来反映由hap i和hap ii得到的dna片段的长度分布中的差异(δq)。δq=q
hapi
–qhapii
,其中q
hapi
为hap i dna片段对应的短片段比例,而q
hapii
为hap ii dna片段对应的短片段的比例。q
hapi
和q
hapii
为两组单倍型各自对应的片段长度分布的统计值的实例。
[0130]
如之前部分所示,当hap ii在肿瘤组织中被删除时,就hap i dna片段而言的长度分布比就hap ii dna片段的长度分布短。结果,观察到正的δq值。可以将正的δq值与阈值相比,从而测定δq是否足够大以断定缺失是存在的。hap i的扩增还显示了正的δq值。当肿瘤组织中存在hap ii的复制时,就hap ii dna片段而言的长度分布小于就hap i dna片段而言的长度分布。因此,δq值称为负的。在没有染色体畸变的情况下,血浆/血清中,就hap i和hap ii dna片段而言的长度分布是相似的。因此,δq值大致为零。
[0131]
可以将患者的δq与正常个体相比较,从而测定所述的值是否正常。此外,可以将患者的δq值与由患有相似癌症的患者得到的值相比较,从而测定所述的值是否异常。此类比较可以涉及与本文所述的阈值相比较。在疾病监测方面,可以在一段时间内连续地监测值δq。δq值的变化可以表明血浆/血清中肿瘤dna的相对百分比浓度的增加或减少。在该技术的所选的实施方案中,肿瘤dna的相对百分比浓度可以与肿瘤的阶段、疾病的预后和进程有关。下文中将更详细地讨论在不同时间点下使用此类实施方案中的测量方法。
[0132]
由短dna片段贡献的总长度的比列的差异
[0133]
在该实施方式中,使用由短dna片段贡献的总长度的比列。计算机系统可以测定样品中一组dna片段的总长度(例如由给定区域的特定单倍型或仅由给定区域得到的片段)。可以选择不同的截断尺寸(w),在某一给定的截断尺寸之下dna片段被定义为“短片段”。可以改变并进行选择不同截断尺寸从而拟合不同的诊断目的。接着,计算机系统可以通过将
等于或小于截断尺寸的随机选择的dna片段的长度加起来来测定短dna片段的总长度。然后,按照如下方式计算由短dna片段贡献的总长度的比列:f=∑wlength/∑nlength,其中∑wlength表示长度等于或小于w(bp)的dna片段的长度的总和;而∑nlength表示等于或小于预定长度n的dna片段的长度的总和。在一个实施方案中,n为600个碱基。但是,其他长度限定,例如150个碱基、180个碱基、200个碱基、250个碱基、300个碱基、400个碱基、500个碱基和700个碱基,都可以用于计算“总长度”。
[0134]
由于illumina genome analyzer系统在扩增和测序长于600个碱基的dna片段中不是非常有效,所以n可以选择600个碱基的值。此外,将所述的分析限定在小于600个碱基的dna片段还可以避免由于基因组结构变异而导致的测量偏差。在存在结构变异的情况下,例如重排(kidd jm et al,nature 2008;453:56-64),以生物信息学为手段将dna片段的末端比对到参照基因组绘制来估计长度时,可能过高估计dna片段的长度。此外,在成功比对到参照基因组的所有dna片段中,大于99.9%的片段均少于600个碱基,因此,包括等于及小于600个碱基的所有片段将提供对样品中的dna片段的长度分布的无偏估计。
[0135]
因此,可以使用由hap i和hap ii之间的短dna片段贡献的总长度的比例的差异。hap i和hap ii dna片段之间的长度分布中的变化可以通过它们的f值反映。在此,我们将f
hap i
和f
hap ii
分别定义为由hap i和hap ii各自所对应的短dna片段的比列。可以按照如下方式计算由hap i和hap ii之间的短dna片段的比例的差异(δf):δf=f
hap i
–fhap ii
。f
hap i
和f
hap ii
为由各个单倍型所对应的两组片段长度分布的统计值。
[0136]
与以上部分所示的实施方案相似,在将hap i dna片段与hap iidna片段相比较时,肿瘤组织中hap ii的删除会导致hap i dna长度分布发生表观上地缩短。这会导致δf为正值。当hap ii被复制时,观察到负的δf值。在缺乏染色体畸变的情况下,δf值接近零。
[0137]
可以将患者的δf与正常个体相比较,从而测定所述的值是否正常。可以将患者的δf与由患有相似癌症的患者得到的值相比较从而测定所述的值是否异常。此类比较可以涉及与本文所述的阈值相比较。在疾病监测方面,可以连续地监测δq值。δf值的变化可以表明血浆/血清中肿瘤dna的百分比浓度的增加或者减少。
[0138]
d.通用方法
[0139]
图10为根据本发明的实施方案,示出用于分析有机体的生物样品的单倍型来测定染色体区域是否表现出缺失或扩增的方法的流程图。生物样品包括源自正常细胞以及潜在的癌症相关细胞的核酸分子(也称为片段)。这些分子在样品中可以是游离的。有机体可以为具有多于一个拷贝的染色体的任何类型,即至少二倍体有机体,但是可以包括更高的多倍体有机体。
[0140]
在本文所述的这种以及任何其他方法的一个实施方案中,生物样品包含游离dna片段。虽然血浆dna的分析用于说明本技术中所述的不同的方法,但是这些方法还可以适用于检测包括正常及肿瘤衍生的dna的混合物的样品中的肿瘤相关的染色体畸变。其他样品类型包括唾液、眼泪、胸膜液、腹水、胆汁、尿、血清、胰液、粪便以及宫颈涂片样品。
[0141]
在步骤1010中,在第一染色体区域处,针对有机体的正常细胞测定第一和第二单倍型。可以通过任何合适的方法,例如本文提及的那些方法,来测定单倍型。可以通过任何方法,例如本文所述的方法,来选择染色体区域。第一染色体区域包括第一多个基因座(例如区域410中的基因座420),而且其是杂合的。杂合性基因座(hets)可以彼此相隔一定距
离,例如在第一多个基因座中,某一基因座可以与另一个基因座相距500个或1000个碱基(或更多)。其他杂合行基因座(hets)可以存在于第一染色体区域中,但是不一定会被利用。
[0142]
在步骤1020中,在生物样品中的多个核酸分子中各个分子具有位置和等位基因的特征。例如,可以鉴别核酸分子在有机体的参照基因组中的位置。可以以多种方式实现该定位,包括利用分子测序(例如通过通用测序),从而得到分子的一个或两个(双末端的)测序的标签,然后将该测序标签与参照基因组比对。可以使用诸如基本局部相似性比对搜索工具(blast)之类的工具来实施此类比对。这种定位可以鉴别为某一染色体臂中的某一个数值。在一个杂合性基因座(hets)处的等位基因可以用于鉴别片段是来自于哪个单倍型。
[0143]
在步骤1030中,基于所鉴别的位置和测定的等位基因,第一组核酸分子被鉴别为得自于第一单倍型。例如,包括图4所示的基因座421(其具有等位基因a)的片段被鉴别为得自hap i。只要第一组核酸分子包括了定位于第一多个基因座中的每个基因座处的至少一个核酸分子,那么其就可以覆盖第一染色体区域。
[0144]
在步骤1040中,基于所鉴别的位置和测定的等位基因,第二组核酸分子被鉴别为得自于第二单倍型。例如,包括图4所示的基因座421(其具有等位基因t)的片段被鉴别为得自hap ii。该第二组包括定位于第一多个基因座中的每个基因座处的至少一个核酸分子。
[0145]
在步骤1050中,计算机系统计算出第一组核酸分子的第一值。该第一值定义了第一组核酸分子的性质。第一值的实例包括在第一组核酸分子数量对应的标签计数以及在第一组核酸分子对应的长度分布。
[0146]
在步骤1060中,计算机系统计算出第二组核酸分子的第二值。第二值定义了第二组核酸分子的性质。
[0147]
在步骤1070中,将第一值与第二值相比较,从而测定第一染色体区域是否表现出缺失或扩增的分类。存在有缺失或扩增的分类可以提供关于有机体具有与癌症相关细胞的信息。比较的实例包括考虑两个值的差异或比值,以及将结果与一个或多个阈值相比较,如本文所述。例如,在sprt分析中,可以将比值与阈值相比较。实例分类可以包括阳性(即所检测到扩增或缺失)、阴性、未确定的以及阳性和阴性对应的变化程度(例如使用1至10的整数,或者0至1的实数)。扩增可以包括简单的复制。此类方法可以检测癌症相关的核酸的存在,其包括肿瘤dna和肿瘤发生前病变(即癌症前体)得到的dna。
[0148]
e.深度
[0149]
分析的深度是指分析所需的分子数量,从而提供达到特定精确度要求的分类或其他测定。在一个实施方案中,可以基于已知的畸变来计算深度,然后在满足该深度的条件进行测量和分析。在另一个实施方案中,可以分析连续进行直到成功分类,并且成功分类对应的深度可以用于测定癌症的水平(例如癌症的阶段或肿瘤的尺寸)。下文提供了与深度有关的一些计算的实例。
[0150]
如本文所述,偏差可以指任何差异或比值。例如,偏差可以介于由阈值或肿瘤浓度得到的第一值和第二值、或第一参数和第二参数之间。如果偏差加倍,则需要测量的片段的数量减少至1/4。更一般性地来讲,如果偏差增大至n倍,则需要测量的片段的数量为原来的1/n2。据此推论,如果偏差减少至1/n,则待检验的片段的数量增大至原来的n2。n可以为实数或整数。
[0151]
假设一种情况,其中肿瘤dna为样品(例如血浆)的10%,并假设测序得到1千万个
片段而且观察到统计学显著性差异。然后,例如,进行富集过程,这样样品中具有20%的肿瘤dna,则所需的片段的数量为2500000个片段。按照这种方式,深度可以与样品中肿瘤dna的百分比浓度相关。
[0152]
此外,扩增的量也影响深度。对拷贝数扩增至原来正常拷贝的两倍(例如4,与正常的2相对)某个区域而言,假设需要x数量的片段进行分析。如果该区域拷贝数增至为原来的4倍,则该区域需要x/4量的片段。
[0153]
f.阈值
[0154]
如上文所述,参数与正常值的偏移量或偏差(例如单倍型之间差值值或比值)可以用于提供诊断。例如,偏差可以为由区域的一个单倍型得到的片段的平均尺寸与由其他单倍型得到的片段的平均尺寸的差异。如果偏差大于某个量(例如由正常样品和/或区域测定的阈值),则缺失或扩增被鉴别到。但是,高于阈值的程度可以是有用的,因此可以使用多个阈值,每个阈值均与癌症的不同水平相应。例如,与正常值的较高的偏差可以提供癌症处于何种分期(例如第4期比第3期具有更高程度的失衡)。此外,较高的偏差还可能是由于肿瘤较大而释放出许多片段、和/或其中所分析的区域被扩增多次。
[0155]
除了提供癌症的不同水平,改变的阈值还允许有效地检测具有畸变的区域或特定的区域。例如,人们可以设定高的阈值,从而主要寻找3倍及更高的扩增,这会得到比一个单倍型的删除更高的失衡。此外,还可以检测2个拷贝的区域的删除。此外,较低的阈值可以用于鉴别可能具有畸变的区域,然后可以对这些区域进行进一步的分析,从而证明畸变是否存在以及存在的位置。例如,可以利用二分法检索(或更高级别的检索,例如八叉树检索),在搜索层次中,较低水平中采用更高的阈值。
[0156]
图11示出了根据本发明的实施方案,在癌细胞中的区域1110中具有缺失畸变的子区域1130以及在血浆中进行的测量从而测定缺失的区域。可以通过本文所提及的任何方法来选择染色体区域1110,例如通过将基因组拆分成相等尺寸的区段中。此外,就基因座1120的每一个基因座而言,图11还示出了等位基因计数的数量。此外,就区域1140(正常区域)和区域1130(缺失的区域)而言,还分别记录累积总数。
[0157]
如果选择区域1110用于分析,hap i的累积计数数量为258,而hap ii的累积计数数量为240,从而在11个基因座上提供的差异为18。此差异占计数总数的百分率比仅分析缺失对应的子区域1130的要小很多。这是合理的,因为在区域1110中大约一半的区域是正常时,而在癌细胞中整的子区域1130都被删除。因此,区域1110中的畸变可能被遗漏,这取决于所用的阈值。
[0158]
为了允许检测子区域的删除,对于相对大的区域而言(就该实例而言,假设区域1110比有待鉴别的删除区域的尺寸相对更大),多个实施方案可以使用较低的阈值。较低的阈值会鉴别更多的区域,其可能包括一些假阳性,但是其会减少假阴性。目前,可以通过进一步的分析来除去假阳性,其还可以精确地定位畸变。
[0159]
一旦区域被标示以用于进一步的分析,则该区域可以分成不同子区域以用于进一步的分析。图11中,人们可以将11个基因座分为两半(例如使用二叉树),从而提供6个基因座的子区域1140和5个基因座的子区域1130。可以使用相同的阈值或更加严谨的阈值来分析这些区域。在该实例中,子区域1140接着被鉴别为正常,而子区域1130被鉴别为包括缺失或扩增。按照这种方式,较大的区域可以被驳回为不具有畸变的结论,并可以花费时间来进
一步分析有可疑的区域(高于下限阈值的区域),从而以高的可靠性(例如使用较高的阈值)去鉴别显示出畸变的子区域。尽管本文使用了rhdo,但是尺寸的技术都是同样适用的。
[0160]
可以基于待检测的及畸变的尺寸来选择用于第一级水平搜索的区域的尺寸(以及在所述的树中较低水平的子区域的尺寸)。已有研究发现癌症显示出10个具有长度为10mb的畸变区域。此外,患者还具有的100mb的畸变区域。较晚期的癌症可以具有更大尺寸的畸变。
[0161]
g.区域内畸变位置的改良
[0162]
在上一个这部分,讨论了基于树搜索的由区域形成子区域的划分。在此,我们讨论用于分析子区域的其他方法,以及如何在区域内精确地定位畸变。
[0163]
图12示出了根据本发明的实施方案,如何使用rhdo分析可以绘制畸变的位置。水平示出染色体区域,其中非癌细胞的单倍型标记为hap i和hap ii。在癌细胞中hap ii的缺失的区域被标记为loh。
[0164]
如所示,rhdo分析由假设染色体区域1202的左侧开始至右侧。每个箭头表示rhdo的分类染色体区段。每个染色体区段都可以被认为是其自己的区域,具体而言为较大杂合子区域的子区域。在分类确定之前,rddo分类节段的尺寸取决于基因座的数量(以及基因座的位置)。在每个rhdo节段中所包括的基因座的数量取决于用于各个节段分析的分子数量、所需的可靠度(例如sprt中的比值比)、以及样品中肿瘤衍生的dna的相对百分比浓度。如图4和图5所示的实例中,当分子的数量足以测定两个单倍型之间存在统计学显著性差异时,则将进行分类。
[0165]
每个实心水平箭头表示rhdo分类节段,其示出在dna样品中不存在单倍型剂量的失衡。在肿瘤中不具有loh的区域中,形成6个rhdo分类,并且每个分类都表示不存在单倍型剂量的失衡。下一个rhdo分类节段1210穿过具有loh的区域和不具有loh的区域之间的结合点1205。在图12的下方部分中,示出了用于rhdo节段1210的sprt曲线。黑色垂直箭头表示具有loh的区域和不具有loh的区域之间的结合点。随着累积从具有loh的区域得到的不断增多数据,该染色体区段的rhdo分类表明存在单倍型剂量的失衡。
[0166]
每个白色的水平箭头表示rhdo分类节段,其表明存在单倍型剂量的失衡。此外,在右侧接下来的4个rhdo表明在dna样品中存在单倍型剂量的失衡。可以推断在具有loh的区域和不具有loh的区域之间的结合点位于第一个显示出rhdo分类变化的区段中,即由存在单倍型剂量的失衡变为缺乏单倍型剂量的失衡,反之亦然。
[0167]
图13示出了根据本发明的实施方案,由另一个方向开始的rhdo的分类。图13中,示出了由两个方向开始的rhdo分类。由左侧开始的rhdo分析,可以推断在具有loh的区域和不具有loh的区域之间的结合点位于第一个显示出存在单倍型剂量失衡的rhdo节段1310内。由右侧开始的rhdo分析,可以推断所述的结合点位于第一个显示出缺乏单倍型剂量失衡的rhdo节段1320内。将由两个方向实施的rhdo分析得到的信息结合起来,可以推断在具有loh的区域和不具有loh的区域之间的结合点位置1330。
[0168]
iv.畸变的非特异单倍型的检测
[0169]
rhdo方法依赖于使用杂合基因座。现在,二倍体有机体的染色体具有一些差异,从而得到两个单倍型,但是杂合基因座的数量是变化的。一些个体可以具有相对较少的杂合基因座。此外,该部分中所述的实施方案还可以用于纯合基因座,其中通过比较两个区域而
非同一区域上的两个单倍型。因此,尽管由于两个不同的染色体区域相比,可能存在一些缺点,但是可以得到更多的数据点,。
[0170]
在相对染色体区域剂量方法中,将由一个染色体区域得到的片段的数量(例如通过计数比对到该区域的测序标签来测定)与期望值(其可以得自参照染色体区域或得自在已知健康的另一个样品中的相同区域)相比较。在这种方式中,可以针对染色体区域而不管测序标签得自哪个单倍型来计数片段。因此,仍可以使用不包含杂合性位点的测序标签。为了实施比较,比较前,实施方案可以将标签计数归一化。通过至少两个基因座定义每个区域(所述的两个基因座彼此分开),并且在这些基因座处的片段可以用于获得关于区域的总值。
[0171]
就特定的区域而言,可以通过将比对到该区域的测序读数的数量除以比对到整个基因组的测序读数的数量来计算测序读数(标签)的归一化值。该归一化的标签计数允许将由一个样品得到的结果与另一个样品的结果相比较。例如,归一化的值可以为由特定区域得到的测序读数的期望比例(例如百分率或分数),如上文所述。但是,许多其他的归一化是可行的,这对于本领域的技术人员而言是显而易见的。例如,人们可以通过将一个区域的计数的数量除以参照区域(在上述情况下,参照区域就是整个基因组)的计数的数量来归一化。然后,可以将该归一化的标签计数与阈值相比较,该阈值可以由未患有癌症的一个或多个参照样品测定得到。
[0172]
接着,将测试个体的归一化标签计数与一个或多个参照受试者(例如未患癌症的)的归一化标签计数相比较。在一种实施方案中,通过针对特定的染色体区域计算测试个体的z分数来进行比较。使用以下等式来计算z分数:z分数=(所述情况下的归一化的标签计数
–
平均值)/s.d.,其中“平均值”为就参照样品而言,比对到特定染色体区域的归一化标签计数的平均值;而s.d.为就参照样品而言,比对到特定区域的归一化标签计数的标准差。因此,z分数为相对于标准差的数量,即,针对某一染色体区域而言,测试个体的归一化标签计数距离参照受试者(一个或多个)的归一化标签计数的平均值多少标准差的数量。
[0173]
在所检验的有机体患有癌症的情况下,在肿瘤组织中扩增的染色体区域在血浆dna中是呈现过量的。这会得到正值的z分数。另一方面,在肿瘤组织中删除的染色体区域在血浆dna中是呈现不足的。这会得到负值的z分数。z分数的量级取决于多种因素。
[0174]
一个因素为在生物样品(例如血浆)中肿瘤衍生的dna的相对百分比浓度。样品(例如血浆)中肿瘤衍生的dna的相对百分比浓度越高,则所测试个体的归一化标签计数与参照个体的计数之间的差异越大。因此,得到较大量级的z分数。
[0175]
另一个因素为在一个或多个参照个体归一化标签计数的波动情况。在所检验个体的生物样品(例如血浆)中,在染色体区域呈现过量的程度相同的情况下,在参照组中归一化标签计数的较小的波动(即,较小的标准差)会得到较大的z分数。类似地,在所检验情况的生物样品(例如血浆)中,在染色体区域的呈现不足的程度相同的情况下,在参照组中归一化标签计数的较小的标准差会得到绝对值更大的负的z分数。
[0176]
另一个因素为肿瘤组织中染色体畸变的程度。就特定的染色体区域而言,染色体畸变的程度是指拷贝数的变化(增加或失去)。在肿瘤组织中拷贝数变化越大,则血浆dna中特定染色体区域的呈现过量或呈现不足的程度越高。例如,两个拷贝的染色体的失去会导致血浆中染色体区域的呈现不足的程度高于两个拷贝的染色体之一的失去所产生的呈现
不足的程度,因此会得到绝对值更大的负的z分数。通常,在癌症中具有多个染色体畸变。在各种癌症中染色体畸变可以因其性质(即扩增或删除)、其程度(单一的或多拷贝的增加或失去)及其长度(畸变的尺寸)的不同而进一步的发生变化。
[0177]
测量归一化标签计数的精确性会受到所分析的分子数量的影响。我们预计在百分比浓度分别为大约12.5%、6.3%和3.2%时,需要分析15,000、60,000和240,000个分子来检测具有一个拷贝变化的染色体畸变(增加或失去)。用于针对不同染色体区域来检测癌症的标签计数的详情在题为“diagnosing fetal chromosomal aneuploidy using massively parallel genomic sequencing”的美国专利公开no.2009/0029377(lo et al.)中有所描述,就所有目的而言,该申请的全部内容以引用方式并入本文。
[0178]
此外,多个实施方案还可以使用长度分析而不用标签计数的方法。此外,还可以使用长度分析而不用归一化的标签计数。如本文所提及以及在美国专利申请no.12/940,992中所述,长度分析可以使用多个参数。例如,可以使用由上文所得的q或f值。由于此类长度值未与读数的数量成比例,所以这些值无需通过由其他区域得到的计数进行归一化。单倍型特异性方法的技术同样也可以应用于非特异的方法。例如,可以使用涉及区域的深度和改良的技术。在一些实施方案中,当比较两个区域时可以考虑根据特定区域的gc含量进行校正。由于rhdo方法使用了相同的区域,所以无需这种校正。
[0179]
v.多个区域
[0180]
尽管某些癌症通常可以与特定染色体区域一起呈现,但是此类癌症并非总是仅在相同的区域中呈现。例如,其他的染色体区域会显示畸变,并且此类其他的区域的位置可能是未知的。此外,当筛查癌症以便鉴别早期癌症时,人们会想鉴别多种癌症,这些癌症在整个基因组中的任何地方都可能呈现畸变。为解决决这些情况,多个实施方案可以用于系统地分析多个区域,从而测定哪个区域显示出畸变。例如,畸变的数量和它们的位置(例如它们是否连续)可以用来确认畸变、癌症的阶段、提供癌症的诊断(例如数量是否大于某个特定阈值)以及基于呈现畸变的多个区域的数量和位置提供预后。
[0181]
因此,多个实施方案可以基于显示畸变的区域的数量来鉴别有机体是否患有癌症。因此,人们可以检验多个区域(例如3000),从而鉴别显示畸变的区域的数量。所述的区域可以覆盖整个基因组或者仅仅覆盖基因组的一部分,例如非重复的区域。
[0182]
图14为根据本发明的实施方案,使用多个染色体区域来分析有机体的生物样品的方法1400的流程图。生物样品包括核酸分子(也称为片段)。
[0183]
在步骤1410中,鉴别有机体的多个非重叠的染色体区域。每个染色体区域都包括多个基因座。如上文提及,区域的尺寸可以为1mb,或者一些其他相等的尺寸。整个基因组则可以包括大约3000个区域,每个区域都具有预定的尺寸和位置。此外,如上文提及,此类预定的区域可以改变,从而容纳特定染色体的特定长度或者待使用的区域的特定数量,以及本文所提及的任何其他标准。如果区域具有不同的长度,可以使用此类长度将结果归一化,例如如本文所述。
[0184]
在步骤1420中,针对多个核酸分子的每一个分子鉴别其在有机体的参照基因组中核酸分子的位置。可以以本文所提及的任何方式测定位置,例如通过测序片段从而获得测序标签并将测序标签与参照基因组比对。此外,分子的特定单倍型可以被检测从而且可以使用单倍型特异的方法中。
[0185]
针对染色体区域的每一个实施步骤1430-1450。在步骤1430中,基于所鉴别的位置,鉴别各组核酸分子所对应的染色体区域。各组核酸分子至少包括位于染色体区域的多个基因座的每个基因座处的一个核酸分子。在一个实施方案中,所述的组可以为比对于染色体区域的特定单倍型的片段,例如,如在上述rhdo方法中所述。在另一个实施方案中,所述的组可以为比对于染色体区域的任何片段,例如在部分iv中所述的方法。
[0186]
在步骤1440中,计算机系统计算出各组核酸分子的各值。各值定义了各组核酸分子的性质。各值可以为本文提及的任何值。例如,所述的值可以为所述的组中的片段的数量或者所述的组中片段的长度分布的统计值。各值还可以为归一化的值,例如区域的标签计数除以该样本的标签计数的总数量,或除以参照区域的标签计数的数量。此外,各值还可以是差值或比值(例如在rhdo中),由此提供不同的区域所对应的性质。
[0187]
在步骤1450中,将各值与参照值相比较,从而测定第一染色体区域是否表现出缺失或扩增的分类。该参照值可以为本文所述的任何阈值或参照值。例如,参照值可以为针对正常样品测定的阈值。对于rhdo而言,各值可以为两种单倍型的标签计数的差异或比值,并且参照值可以为用于测定统计学上显著性差异存在的阈值。例如,参照值可以为另一个单倍型或区域的标签计数或尺寸,并且比较可以包括但不局限于差值或比值(或此类值的函数),然后测定差值或比值是否大于阈值。
[0188]
参照值可以基于其他区域的结果而改变。例如,如果相邻区域也显示出偏差(尽管与一个阈值相比,显得较小,例如z分数为3),但是可以使用较低阈值。例如,如果3个连续的区域均高于第一阈值,则患癌症可能性更大。因此,该第一阈值可以低于另一个由非连续的区域来鉴别癌症所必需的阈值。具有较小的偏差3个区域(或多于3个区域)可以使随机波动的影响的可能性降到足够地低,如此一来可以保持较高敏感性和特异性。
[0189]
在步骤1460中,测定被分类为表现出缺失或扩增的染色体区域的数量。对所计数的染色体区域可以具有有一些限制。例如,可以只计数至少与一个其他区域相连续的区域(或者可以要求连续的区域达到某一尺寸,例如4或更多的区域)。就其中区域并非相等的实施方案而言,数量可以考虑各自不同的长度(例如数量可以为畸变区域的总长度)。
[0190]
在步骤1470中,将量与量的阈值相比较,从而判断样品的分类。例如,分类可以为有机体是否患有癌症、癌症的阶段以及癌症的预后。在一个实施方案中,计数所有的畸变区域,并使用单一的阈值,而不管区域在何处出现。在另一个实施方案中,基于所计数的区域的位置和尺寸,阈值可以改变。例如,可以将特定染色体或染色体臂上的区域的量与该特定染色体(或臂)的阈值相比较。可以使用多个阈值。例如,在特定染色体(或臂)上的畸变区域的量必须大于第一阈值,并且基因组中畸变区域的总量必须大于第二阈值。
[0191]
这种用于区域的量的阈值还可以取决于所计数区域的失衡有多强。例如,测定癌症分类的阈值的区域的量可以取决于测定各区域中的畸变的特异性和敏感性(畸变阈值)。例如,如果畸变阈值低(例如z分数为2),则可以选择高的量的阈值(例如150)。但是,如果畸变阈值高(例如z分数为3),则量的阈值可以较低(例如50)。此外,显示畸变的区域的量还可以为加权值,例如相比于显示较少失衡的区域,可以给显示高度失衡的区域赋予较高权重(即存在比仅为畸变阳性和阴性更多的分类)。
[0192]
因此,染色体区域的量(其可以包括数量和/或尺寸)可以用于反映疾病的严重性,其中所述的染色体区域指的是对应的归一化标签计数(或者其他组别的属性对应的各值)
展现出显著地呈现过量或呈现不足。具有由归一化标签计数显示畸变的染色体区域的量可以通过两个因子来测定,即肿瘤组织中染色体畸变的数量(或尺寸),以及生物样品中肿瘤衍生的dna的百分比浓度(例如血浆)。更加晚期的癌症往往显示更多(以及更大)的染色体畸变。因此,与癌症相关的较大的染色体畸变是可潜在地被检测到。在患有更晚期的癌症的患者中,较高的肿瘤负荷会导致血浆中肿瘤衍生的dna的百分比浓度更高。结果,在血浆样品中,肿瘤相关的染色体畸变得更容易被检测到。
[0193]
在癌症筛查或检测方面,染色体区域的量可以用于测定所检验的受试者患有癌症的可能性,其中所述的染色体区域指的是该区域对应的归一化标签计数(或其他值)展现出显著地呈现过量或呈现不足。使用
±
2的截断(即,z分数》2或《-2),预计大约5%的检验区域的z分数会由于随机波动的偶然性而显示其与对照受试者的平均值存在显著性偏差。当将整个基因组分为1mb片段时,整个基因组存在大约3000个染色体区段。因此,预计大约150个染色体区段具有》2或《-2的z分数(由于随机波动导致)。
[0194]
因此,对于z分数为》2或《-2的区段数量而言,阈值为150可以用于测定癌症是否存在。对于具有畸变z分数的区段数量(例如100、125、175、200、250和300)而言,可以选择其他截断值来拟合诊断目的。较低的截断值(例如100)会得到更高的敏感性检验但较低的特异性,而较高的截断值会得到更高的特异性但较低的敏感性。可以通过增大z分数的截断值来减少假阳性分类的数量。例如,如果截断值增至3,则仅0.3%的区段为假阳性。在这种情况下,3个以上的区段具有畸变z分数可以用于表明癌症的存在。此外,可以选择其他阈值,例如1、2、4、5、10、20和30,从而拟合不同的诊断目的。但是,随着进行诊断所需的畸变片段的数量的增加,检测癌症相关的染色体畸变的敏感性会降低。
[0195]
用于改善敏感性但不会牺牲特异性的一种可行的方法将考虑相邻染色体区段的结果。在一个实施方案中,用于z分数的截断值保持为》2和《-2。但是,仅当两个连续的区段显示出相同类型的畸变时,例如两个区段都具有》2的z分数,则染色体区域将被分类为潜在的畸变。如果归一化标签计数的偏差为随机误差,则具有两个连续区段(在同一方向为假阳性)的可能性为0.125%(5%x5%/2)。另一方面,如果染色体畸变涵盖了两个连续的区段,较低的截断值将使得对血浆样品中区段的过多呈现或呈现不足的检测更敏感。由于归一化的标签计数(或其他值)与对照受试者的平均值的偏差并非由于随机误差,所以连续分类的要求不会对敏感性具有显著不利的影响。在其他实施方案中,如果使用较高的截断值,可以将相邻区段的z分数加在一起。例如,可以将3个连续片段的z分数合计,并可以使用截断值为5。这种概念可以扩展至多于3个连续的区段。
[0196]
此外,数量和畸变阈值的组合还可以取决于分析的目的以及对有机体的任何的先验知识(或对其缺乏了解)。例如,如果针对正常的健康群体进行癌症筛查,则通常使用高特异性对应的区域的数量(即就区域的数量而言使用的高阈值)和畸变阈值去识别某个区域是否具有染色体畸变。但是,在具有较高的风险的患者(例如有肿块或家族史、吸烟、hpv病毒、肝炎病毒或其他病毒的患者)中,则可以降低阈值,从而具有更高的敏感性(较低的假阴性)。
[0197]
在一个实施方案中,如果使用1mb分辨率和检测极限为6.3%的肿瘤衍生dna来检测染色体畸变,则每个1mb区段中分子的数量必须为60000。对于整个基因组而言将,则需要大约180000000(60000读数/mb x 3000mb)个可比对的读数。
[0198]
图15示出了根据本发明的实施方案,在表格1500中阐明不同数量的染色体区段以及肿瘤衍生的片段的相对百分比浓度所需的深度。列1510提供了由样品的肿瘤细胞得到的dna片段浓度。浓度越高,越容易检测畸变,因此需要用于分析的分子数量也越少。列1520提供了每个染色体区段所需的分子的评估数量,该数量可以通过上文关于深度的部分中所描述的方法来计算。
[0199]
较小尺寸的染色体区段会得到较高的分辨率,适用于检测较小的染色体畸变。但是,这会需要增加用于分析的分子数量。在损失分辨率的代价下,较大尺寸的染色体区段会减少用于分析所需的分子数量。因此,仅可以检测较大的畸变。在一个实施方式中,使用的区域越大,则显示畸变的染色体区段将被细分并对这些子区域进行分析,从而得到较高的分辨率(例如上文所述)。列1530提供了各个染色体区段的尺寸。值越小,则对应的区域数目越多。列1540示出了就整个基因组而言,所需要的分子数量。因此,如果人们具有上文所述的评估参数(或最小的检测浓度),则可以确定需要用于分析的分子数量。
[0200]
vi.在一定时间内的进程
[0201]
随着肿瘤发生的进程,由于肿瘤将释放更多的dna片段(例如由于肿瘤的生长、更多的坏疽或更高的血管分布),则血浆中的肿瘤dna片段的量将增多。由肿瘤组织对应的较多的dna片段进入到血浆中将增加血浆中失衡的程度(例如在rhdo中,两种单倍型之间的标签计数的差异将增加)。此外,由于肿瘤dna片段的数量增加,则其中存在畸变的区域的数量可以更容易地被检测到。例如,区域中的肿瘤dna的量太少以至于染色体畸变不能被检查到。其原因在于在肿瘤较小并只释放少量的癌症dna片段情况下,没有足够的片段进行分析,所以不能建立统计学显著性差异,使得畸变不能被检测。即使在肿瘤较小时,也可能得到较多的片段用于分析,但是这可能需要大量的样品(例如大量的血浆)。
[0202]
跟踪癌症的进程可以使用一个或多个区域中畸变的量(例如通过失衡或所需的深度),或者表现出畸变的染色体区域的量(数量和/或尺寸)。在一个实例中,如果一个区域(或多个区域)的畸变的量比其他区域畸变的量增加的更快,则该区域可以用作优选的分子标志来检测癌症。这种增加可能是由于肿瘤变大和/或区域被多次扩增而释放出更多的dna片段的结果,。此外,人们还可以监测术后畸变值(例如畸变的量或显示畸变的区域的数量,或者它们的组合)的变化情况来确认肿瘤是否被完全切除。
[0203]
在所述技术的多种实施方式中,测定肿瘤dna的相对百分比浓度可用于癌症分期、预后或监测癌症的进程。所测得的进程可以提供关于癌症目前的癌症分期和癌症多快地生长或扩散的信息。癌症的“分期”与以下因素的全部或一部分有关:肿瘤的尺寸、组织学表现、淋巴结转移存在/缺乏以及远端转移存在/缺失。癌症的“预后”涉及评估疾病进程的概率和/或由癌症存活的概率。此外,其还涉及时间的评估,其间患者可能不具有临床演变或存活时间。癌症的“监测”涉及查看癌症是否进行(例如尺寸增大、淋巴结转移、或蔓延至远端器官,即,转移)。此外,监测还可以涉及检查肿瘤是否被治疗控制。例如,如果治疗是有效的,则人们可能会看到肿瘤尺寸的减小,转移或淋巴结转移的消退、患者的全面健康的改善(例如体重增加)。
[0204]
a.癌症dna的相对百分比浓度的测定
[0205]
其中一种跟踪一个或多个区域的畸变的量增加的方法为测定该区域的癌症dna的相对百分比浓度。然后,可以使用癌症dna的相对百分比浓度的变化在一定时间内跟踪肿
瘤。这种跟踪可以用于诊断,例如第一测量可以提供背景水平(其可以对应于人的一般畸变水平),而后来的测量如果可以看到变化,其表明肿瘤生长(因此为癌症)。此外,癌症dna的相对百分比浓度的变化可以用于评价治疗的预后效果。在所述技术的其他实施方案中,血浆中肿瘤dna的相对百分比浓度的增加表明较差的预后,或者患者的肿瘤负荷增大。
[0206]
可以以多种方式测定癌症dna的相对百分比浓度。例如,在标签计数中,一个单倍型与另一个单倍型的差异(或者一个区域与另一个区域相比)。另一种方法为见在到统计学显著性差异之前的深度(即,所分析的片段的数量)。前者的实例,单倍型剂量的差异可以用于通过分析具有杂合性缺失的染色体区域来测定生物样品(例如血浆)中肿瘤衍生的dna的相对百分比浓度。
[0207]
已有研究显示肿瘤衍生的dna的量与癌症患者中的肿瘤负荷呈正相关(lo et al.cancer res.1999;59:5452-5.和chan et al.clin chem.2005;51:2192-5)。因此,通过rhdo分析来连续监测生物样品(例如血浆样品)中肿瘤衍生的dna的相对百分比浓度可以用于监测患者的疾病的进程。例如,在治疗后连续收集的样品(例如血浆)中,肿瘤衍生的dna的相对百分比浓度的监测可以用于测定治疗的成功情况。
[0208]
图16示出了根据本发明的实施方案,通过rhdo分析测量血浆中肿瘤衍生的dna的相对百分比浓度的原理。测定两种单倍型之间的失衡,并且失衡的程度可以用于测定样品中肿瘤dna的相对百分比浓度浓度。
[0209]
hap i和hap ii表示非肿瘤组织中的两种单倍型。hap ii在肿瘤组织中在子区域1610中被部分删除。因此,在血浆中检测的与删除的区域1610相应的hap ii相关片段是由非肿瘤组织贡献的。另一方面,hap i中的区域1610在肿瘤和非肿瘤组织中呈现。因此,hap i和hap ii的读数计数之间的差异表示血浆中肿瘤衍生的dna的量。
[0210]
可以使用以下公式,对于受到loh影响的染色体区域,由删除的及非删除的染色体得到的测序读数(标签)的数量来计算肿瘤衍生的dna的相对百分比浓度:f=(n
hapi
–nhapii
)/n
hapi x 100%,其中n
hapi
为就位于受到loh影响的染色体区域中的杂合snp而言,与在hap i上的等位基因相应的测序读数的数量;而n
hapii
为就位于受到loh影响的染色体区域1610中的杂合snp而言,与在hap ii上的等位基因相应的测序读数的数量。
[0211]
上述公式相当于定义p作为位于不包括删除的染色体区域(hap i)上的杂合基因座的累积标签计数,并定义q作为位于包括删除1610的染色体区域(hap ii)上的累积标签计数,并且样品中肿瘤dna的相对百分比浓度(f)计算为f=1-q/p。如图11所示的实例,肿瘤dna的相对百分比浓度为14%(1
–
104/121)。
[0212]
在肿瘤切除前和切除后收集hcc患者中一定肿瘤dna百分比浓度的血浆样品a。在肿瘤切除前,就给定染色体区域的第一单倍型,n
hapi
为30443,而对于染色体区域的第二单倍型,n
hapii
为16221,其得到f为46.7%。在肿瘤切除后,n
hapi
为31534,而n
hapii
为31098,其得到f为1.4%。这种监测显示肿瘤切除是成功的。
[0213]
游离dna尺寸分布的变化程度还可以用于测定肿瘤dna相对百分比浓度。在一个实施方式中,可以测定血浆中源自肿瘤和非肿瘤组织dna的对应的确切尺寸分布,然后所测量的落入两个已知的分布之间的尺寸分布可以提供肿瘤dna百分比浓度(例如使用肿瘤和非肿瘤组织的尺寸分布的两个统计值之间的线性模型)。此外,也可以使用尺寸变化进行连续监测。在一个方面中,尺寸分布的变化被测定为与血浆中肿瘤dna百分比浓度成比例。
[0214]
此外,还可以以相似的方式使用不同区域之间的差异,即,上文所述的非特异的单倍型检测方法。在标签计数方法中,多个参数用于监测疾病的进程。例如,就显示染色体畸变的区域而言,z分数的量级可以用于反映生物样品(例如血浆)中肿瘤衍生的dna的百分比浓度。特定区域的过多呈现或呈现不足的程度与样品中肿瘤衍生的dna的百分比浓度或肿瘤组织中拷贝数变化的程度或数量成比例。z分数的量级是度量样品中特定染色体区域与对照受试者相比的过多呈现或呈现不足的程度的参数。因此,z分数的量级可以反映样品中肿瘤dna相对百分比浓度,并由此反映患者的肿瘤负荷。
[0215]
b.跟踪区域的数量
[0216]
如上文所提及,表现出染色体畸变的区域的数量可以用于筛选癌症,并且还可以用于监测和预后。例如,如果癌症复发或者如果治疗起作用,则所述的监测可以用于测定癌症的当前阶段的状况。当肿瘤在进程中时,肿瘤的基因组构成将更多被降解。为了鉴别这种连续的降解,跟踪区域(例如1mb的预定区域)的数量的方法可以用于鉴别肿瘤的进程。在更加晚期的癌症中,肿瘤则会具有表现出畸变的更多的区域。
[0217]
c.方法
[0218]
图17为根据本发明的实施方案,示出使用包括核酸分子的生物样品来测定有机体中染色体畸变的进程的方法的流程图。在一个实施方案中,至少一些核酸分子是流离的。例如,染色体畸变可能来自于恶性肿瘤或恶化前的病变。此外,畸变的增多可能是由于在一定时间内有机体具有染色体畸变的细胞越来越多,或者由于有机体具有一定比例的细胞其对应的每个细胞的畸变量增多。作为减少的实例,治疗(例如手术或化疗)可以除去或减少与癌症相关的细胞。
[0219]
在步骤1710中,鉴别有机体的一个或多个非重叠的染色体区域。各个染色体区域包括多个基因座。可以通过任何合适的方法鉴别区域,例如本文所述的那些。
[0220]
在多个时间点下的每个时间实施步骤1720-1750。每个时间点对应于在由有机体获得样品时的不同时间。当前的样品为在给定时间期间进行分析的样品。例如,可以在6个月中的每个月取一次样品,并可以在获得样品后尽快进行分析。除此之外,可以在跨越多次时间期间取得多次测量后进行分析。
[0221]
在步骤1720中,分析有机体的当前生物样品,从而鉴别核酸分子在有机体的参照基因组中的位置。可以以本文所提及的任何方式测定位置,例如通过对片段进行测序来获得测序的标签,并将测序标签与参照基因组比对。此外,还可以针对基于单倍型特异性的方法,测定分子的特定单倍型。
[0222]
针对一个或多个染色体区域的每一个区域实施步骤1730-1750。当使用多个区域时,可以使用由部分v得到的实施方案。在步骤1730中,基于所鉴别的位置,可鉴别各组核酸分子所对应的染色体区域。各组核酸包括位于所述的染色体区域中的多个基因座中每一个基因座处的至少一个核酸分子。在一个实施方案中,所述的组可以为比对到染色体区域的特定单倍型的片段,例如在上文rhdo方法中所述。在另一个实施方案中,所述的组可以为比对到染色体区域的任何片段,如在部分iv中所述的方法。
[0223]
在步骤1740中,计算机系统计算出各组核酸分子的各值。各值定义了各组核酸分子的性质。各值可以为本文所提及的任意值。例如,所述的值可以为所述的组中的片段的数量,或者为所述的组中的片段的尺寸分布的统计值。此外,各值还可以为归一化的值,例如
对于样品而言,区域的标签计数除以标签计数的总数量,或者除以参照区域的标签计数的数量。此外,各值可以为与另一个值的差值或比值(例如在rhdo中),由此就所述的区域提供差异的性质。
[0224]
在步骤1750中,将各值与参照值相比较,从而测定第一染色体区域是否表现缺失或扩增的分类。该参照值可以为本文所述的任何阈值或参照值。例如,参照值可以为针对正常样品测定的阈值。对于rhdo,各值可以为两种单倍型的标签计数的差值或比值,并且参照值可以为用于测定统计学显著性差异存在的阈值。作为另一个实例,参照值可以为另一个单倍型或区域的标签计数或尺寸,并且比较可以包括但不局限于差值或比值(或此类值的函数),然后测定差值或比值是否大于阈值。可以根据任何合适的方法和标准测定参照值,例如本文所述。
[0225]
在步骤1760中,在多个时间点下各个染色体区域的分类用于测定有机体中染色体畸变的性质。所述的进程可以用于测定有机体是否患有癌症、癌症分期以及癌症的预后。每个这些测定可以涉及癌症的分类,如本文所述。
[0226]
可以以多种方式实施这种癌症的分类。例如,可以计数畸变区域的量,并与阈值相比较。就区域而言,分类可以为数值(例如肿瘤的浓度,并且参照值可以为用于不同单倍型或不同区域的值),并且可以测定浓度的变化。可以将浓度的变化与阈值相比较,如果测定到显著增多的情况发生,由此表示有肿瘤存在的信号。
[0227]
vii.实施例
[0228]
a.使用基于sprt的rhdo
[0229]
在该部分中,我们示出了针对肝细胞癌(hcc)患者,使用sprt的相对单倍型剂量(rhdo)来分析的实例。在该患者的肿瘤组织中,观察到两个4号染色体中的一个被删除。这会导致4号染色体上snp的杂合性缺失。就患者的单倍型而言,分析患者、其妻子及儿子的基因组dna,并测定3位个体的基因型。患者的结构性单倍型由它们的基因型推导得到。实施大规模平行测序,并鉴别和计数与4号染色体的两个单倍型相应的、具有snp等位基因的测序读数。
[0230]
上文已经描述了rhdo和sprt的等式和原理。在一个实施方案中,rhdo分析可以通过计算机编程的手段去检测例如在dna样品中单倍型剂量的10%的差异,其中当两个单倍型中的一个单倍型被扩增或删除时,所述的单倍型的剂量差异与肿瘤dna浓度10%相对应。在其他实施方案中,可以设定rhdo分析的敏感性,从而检测dna样品中2%,5%,15%,20%,25%,30%,40%和50%的肿瘤衍生的dna。利用计算sprt分类区域的上限阈值和下限阈值的参数,可以调节rhdo分析的敏感性。例如使用比值比(一个单倍型的标签计数相对于其他单倍型的标签计数的比值),可调节的参数可以为所需的检测极限水平(例如多少百分率的肿瘤浓度应该是可检测的,其将影响所分析的分子的数量)以及用于分类的阈值。
[0231]
在该rhdo分析中,零假设为4号染色体的两个单倍型以相同的剂量存在。备择假设为生物样品(例如血浆)中,两个单倍型的剂量相差多于10%。针对两种假设,具有snp等位基因(与两个单倍型相应)的测序读数的数量以由不同的snp累积得到的数据形式用统计学的方式去比较。当累积的数据足以测定两个单倍型剂量是否以等量存在或者在统计学上相差至少10%时,进行sprt分类。在4号染色体的q臂上,典型的sprt分类区域示于图18a中。阈值10%在此仅用于示例说明的目的。此外,还可以检测其他程度的差异(例如0.1%,1%,
cytogenetics in oncology and haematology(atlasgeneticsoncology.org//tumors/tumorliste.html))。相反,在非靶向全基因组方法中,不可能发生染色体畸变的区域与具有潜在的畸变的区域都是进行相同的程度(深度)的分析。
[0237]
我们使用靶向分析策略来分析由3位hcc患者和4位健康对照受试者得到的血浆样品。使用得自agilent的sureselect捕获系统来实现靶标的富集(gnirke et al.nat.biotechnol 2009.27:182-9)。选择sureselect系统作为一种可行的靶标富集技术的实例。其他液相(illuminatruseq custom enrichment system)或固相(例如roche-nimblegen system)靶标捕获系统以及基于扩增子的靶标富集系统(例如quantalifesystem和raindance system)也可以使用。设计捕获探针,使其位于在hcc中普通的及罕见的显示畸变的染色体区域上。在靶标捕获后,则在illuminagaiix分析仪的流通池(flowcell)中以一个通道(lane)对应一个dna样品的形式进行测序。极低概率发生扩增和删除的区域被用作参照以便与其中相对经常地存在扩增和删除的区域相比较。
[0238]
在图19中,显示出在hcc中发现的普通的染色体畸变(该图改编自wong et al(am j pathol 1999;154:37-43))。在染色体模式图的右侧上的线表示染色体增加,而左侧上的线表示各个患者样品的染色体缺失。粗线表示高水平的增加。长方形表示靶标捕获探针的位置。
[0239]
靶向标签计数分析
[0240]
就染色体畸变的检测而言,对于潜在的畸变区域和参照区域,我们首先计算归一化的标签计数。接着,如之前chen et al(plos one 2011;6:e21791)所述,针对区域的gc含量校正归一化的标签计数。在当前的实例中,选择8号染色体的p臂作为潜在的畸变区域,选择9号染色体的q臂为参照区域。使用affymetrixsnp 6.0微阵列,针对染色体畸变来分析3位hcc患者的肿瘤组织。3位患者的肿瘤组织中,8p和9q的染色体剂量的变化如下所示。患者hcc013的8p减少,而9q无变化。患者hcc027的8p增多,而9q无变化。患者hcc023的8p减少,而9q无变化。
[0241]
接着,使用靶向分析,对3位hcc患者和4位健康对照受试者计算chr 8p和9q之间的归一化标签计数的比值。图20a示出了hcc和健康患者的归一化标签计数比值的结果。对于hcc013和hcc023的实例而言,观察到8p和9q之间的归一化标签计数比例的减小。这与所发现的肿瘤组织中染色体8p的缺失一致。对于hcc027的实例而言,观察到比值增大,并且该增大的比值与这种情况的肿瘤组织中染色体8p的增加一致。虚线分别表示相对于4个正常对照受试者的平均值偏移正负两个标准差。
[0242]
靶向尺寸分析
[0243]
在之前的部分中,我们描述了通过测定癌症患者中血浆dna片段的尺寸分布来鉴别癌症相关改变的原理。此外,可以使用靶标富集方法来检测尺寸的改变。对于3个hcc实例(hcc 013、hcc027和hcc023),在将测序读数与人类参考基因组比对之后测定各测序dna片段的尺寸。通过两个末端最外侧的核苷酸的坐标来推导测序dna片段的尺寸。在其他实施方案中,测序整条dna片段的全长,然后可以由测序长度直接测定片段的尺寸。比较比对到染色体8p的dna片段的尺寸分布与比对到染色体9q的dna片段的尺寸分布。就对两个群体dna的尺寸分布的差异进行检测而言,首先在当前的实例中对各个群体测定短于150bp的dna片段的比例。在其他实施方案中,可以使用其他截断值,例如80bp,110bp,100bp,110bp,
120bp,130bp,140bp,160bp和170bp。δq=q
8p
–q9q
,其中q
8p
为比对到染色体8p且短于150bp的dna片段的比例;而q
9q
为比对到染色体9q且短于150bp的dna片段的比例。
[0244]
由于较短的dna片段尺寸分布会得到较高短于截断值的比例(在当前的实例中即为短于150bp的比列),较高(更大的正值)的δf值表示比对到染色体8p的dna片段相对于比对到染色体9q的那些dna片段具有更短的分布。相比之下,较小(或绝对值更大的负值)的结果表明比对到染色体8p的dna片段相对于比对到染色体9q的那些dna片段具有更长的分布。
[0245]
图20b示出了针对3位hcc患者和4位健康对照受试者在靶标富集及大规模平行测序之后得到的尺寸分析的结果。在4位健康的对照受试者中,正值的δq表明比对到染色体8p的dna片段比比对到染色体9q的那些dna片段具有稍短的尺寸分布。虚线表示针对4位对照受试者,由偏离平均值2个标准偏差内得到的δq所对应的间隔。cases hcc 013和hcc 023的δq值相对于对照受试者的平均值往下偏移大于两个标准偏差。这两种情况在肿瘤组织中具有染色体8p的缺失。就该染色体区域而言,在肿瘤中8p的缺失会导致肿瘤衍生的dna对血浆的贡献减少。由于在外周血中,肿瘤衍生的dna比由非肿瘤组织衍生得到的dna更短,所以这会导致比对到染色体8p的血浆dna片段的尺寸分布明显更长。这与这两个实例中较低(更大的负值)的δq值一致。相比之下,在hcc027情况中8p的扩增会导致与该区域比对的dna片段的分布明显更短。因此,认为比对到8p的血浆dna较高比例的片段是相对比较短的。这与所观察的情况一致,即hcc027的δq值比健康对照受试者具有绝对值更大的正值。
[0246]
c.用于检测肿瘤衍生的染色体畸变的多个区域
[0247]
在肿瘤组织中通常检测到染色体畸变,包括某些染色体区域的删除和扩增。在不同类型的癌症中观察到染色体畸变的特征模式。在此,我们使用多个实例来说明用于检测癌症患者血浆中这些癌症相关的染色体畸变的不同方法。此外,我们的方法还适合用于癌症的筛查、疾病进程的监测以及对治疗的反应。对一位hcc患者及两位鼻咽癌(npc)患者得到的样品进行分析。对于hcc患者,在手术切除肿瘤之前和之后收集静脉血样品。对于两位npc患者,在诊断时收集静脉血样品。此外,对一位慢性肝炎b携带者以及一位在血浆中可检测epstein-barr病毒dna的受试者的血浆样品进行分析。这两位受试者未患有任何癌症。
[0248]
使用微阵列分析来实施对肿瘤衍生的染色体畸变的检测。具体而言,使用affymetrix snp6.0微阵列系统分析由hcc患者的血细胞以及肿瘤样品提取的dna。使用affymetrix genotyping console v4.0测定血细胞和肿瘤组织的基因型。使用birdseed v2算法基于微阵列上snp的不同等位基因的信号强度以及拷贝数改变(cnv)探针来测定染色体畸变,包括增加和删除。
[0249]
基于计数的分析
[0250]
为了在血浆中实施测序标签计数分析,由各受试者收集10毫升静脉血。对各血液样品,在将样品离心后收集血浆。使用qiamp blood mini kit(qiagen)由4-6ml血浆提取dna。按照之前所述构建血浆dna文库(lo ymd.sci transl med 2010,2:61ra91),然后使用illumina genome analyzer平台对该文库进行大规模平行测序。对血浆dna分析实施末端配对的测序。在两个末端的每个末端处对每个分子进行测序(50bp),因此每个分子总计100bp。使用soap2程序(soap.genomics.org.cn/)(li r et al.bioinformatics 2009,25:1966-7),将每个序列的两个末端与人类基因组比对(由ucsc genome.ucsc.edu下载的hg18 ncbi.36)。
[0251]
然后,将基因组分为多个1兆碱基(1mb)染色体区段,并测定比对到各1mb染色体区段的测序读数的数量。接着,根据各染色体区段的gc含量,使用基于局部加权回归散点平滑法(loess)的算法来校正各区段的标签计数(chen e et al.plos one 2011,6:e21791)。该校正的目的在于使与测序相关的定量偏倚(bias)最小,其中所述的定量偏倚是由于在不同基因组区段之间gc含量的差异引起。上文提及的分隔成1mb区段是用于示例的目的。其他节段尺寸也可以使用,例如2mb,10mb,25mb,50mb等。此外,还可以基于特定患者的特定肿瘤以及一般肿瘤的特定类型的基因组特征来选择区段尺寸。此外,例如就单一的分子测序技术而言,如果测序方法可以显示具有较低的gc偏倚,例如helicos system(www.helicosbio.com)或pacific biosciences single molecular real-time system(www.pacificbiosciences.com),则可以省略gc校正步骤。
[0252]
在之前的研究中,我们对得自未患有癌症的受试者的57个血浆样品进行测序。这些血浆测序的结果用于测定各1mb区段的标签计数的参照范围。对于各1mb区段而言,测定57位个体的标签计数的平均值和标准偏差。接着,研究受试者的结果表示为z分数,其是使用以下等式计算的:z-分数=(所述情况的测序标签的量
–
平均值)/s.d,其中平均数值为对于参照样品而言,为比对到特定的1mb区段的测序标签的平均数值;s.d而为就参照样品而言,为比对到特定的1mb节段的测序标签的标准差。
[0253]
图21-24示出了4位研究受试者的测序标签计数分析的结果。在图的边缘示出1mb区段。以顺时针方向自pter-qter来来调整染色体编号及染色体模式图(最外圈)(着丝粒以黄色示出)。在图21中,内圈2101示出通过分析肿瘤而测定的畸变(删除或扩增)的区域。内圈2101以5个级别示出。该级别由-2(最内侧的线)至+2(最外侧的线)。-2值表示就相应区域而言,两个染色体拷贝的损失。对某个染色体区域而言,-1值表示缺失了两个染色体拷贝中的一个。0值表示不具有染色体增加或缺失。+1值表示增加了一个染色体拷贝,而+2表示增加了两个染色体拷贝。
[0254]
中圈2102示出了血浆分析的结果。如同人们可以看见的那样,中圈2102对应治疗前血浆中的畸变结果与内圈的畸变模式相吻合。中圈2102为更多的级别线,但是进程是相同的。外圈2103示出了在治疗后由分析血浆得到的数据点,并且这些数据点是灰色的(证明无过多呈现/呈现不足-无畸变)。
[0255]
在血浆中测序标签过多呈现的染色体区域(z分数》3)由绿点2110表示。在血浆中测序标签呈现不足的区域(z分数《-3)由红点2120表示。在血浆中检测的无显著的染色体畸变的区域(z分数在-3至3之间)由灰点表示。将过多呈现/呈现不足对计数的总数量归一化。在测序前涉及pcr扩增的情况下,归一化可以考虑gc偏倚的校正。
[0256]
图21示出了根据本发明的实施方案的hcc患者的circos图,其描绘了由血浆dna的测序标签的计数得到的数据。由内至外的圈图分别表示:通过微阵列分析检测肿瘤组织的染色体畸变(红色和绿色分别表示删除和扩增);在手术切除肿瘤之前获得的血浆样品的z分数分析;以及在切除之后1个月时获得的血浆样品的z分数分析。在肿瘤切除前,在血浆中检测到的染色体畸变恰好与通过微阵列分析在肿瘤组织中鉴别到的那些畸变相吻合。在肿瘤切除后,大部分的癌症相关的染色体畸变在血浆中消失。这些数据反映了此类方法用于检测疾病的进程和治疗效果的价值。
[0257]
图22示出了根据本发明的实施方案,针对未患有hcc的慢性hbv携带者的血浆样品
所进行的测序标签的计数分析。与hcc患者(图21)相反,癌症相关的染色体畸变在该hbv携带者的血浆中未检测到。这些数据反映了该方法用于癌症筛选、诊断和监测的价值。
[0258]
图23示出了根据本发明的实施方案,针对患有第三期npc的癌症患者的血浆样品所进行的测序标签的计数分析。在治疗前取得的血浆样品中检测到染色体畸变。具体而言,在染色体1,3,7,9和14中鉴别到显著的畸变。
[0259]
图24示出了根据本发明的实施方案,针对患第四期npc的癌症患者的血浆样品所进行的测序标签的计数分析。在治疗前取得的血浆样品中检测到染色体畸变。当与患第三期癌症疾病(图23)的患者相比较时,检测到更多的染色体畸变。此外,测序标签计数与对照平均值偏差的更多,即z分数与0偏差更多(正的或负的)。染色体畸变数量的增多以及测序标签计数与对照相比偏差的程度更高反映了在晚期疾病中基因组改变的程度更严重,因此反映了此类方法用于分期、预后和监测癌症的价值。
[0260]
基于尺寸的分析
[0261]
在之前的研究中,已经显示由肿瘤组织衍生的dna的尺寸分布比由非肿瘤组织衍生的dna的尺寸分布更短(diehl f et al.proc natl acad sci usa 2005,102(45):16368-73)。在之前的研究中,我们概括出通过血浆dna的尺寸分析来检测血浆单倍型失衡的方法。在此,我们使用hcc患者的测序数据来进一步说明该方法。
[0262]
为了达到说明的目的,我们鉴别两个区域用于尺寸分析。在一个区域(1号染色体(chr1);坐标:159,935,347 to 167,219,158)中,在肿瘤组织中检测到两个同源染色体中的一个同源染色体被复制。在其他区域(10号染色体(chr10);坐标:100,137,050至101,907,356)中,在肿瘤组织中检测到两个同源染色体中的一个同源染色体被删除(即loh)。除了测定测序片段来自哪个单倍型,还要用测序片段在参照基因组中的最外侧核苷酸的坐标来以生物信息学方式测定测序片段的尺寸。接着,测定由两个单倍型中的每一个单倍型对应片段的尺寸分布。
[0263]
对于chr10的loh区域,在肿瘤组织中检测到一个单倍型(即删除的单倍型)。因此,比对到该删除的单倍型的所有血浆dna片段均衍生自非癌症组织。另一方面,比对到在肿瘤组织中未被删除的单倍型(非删除的单倍型)的片段可以衍生自肿瘤或非肿瘤组织。由于肿瘤衍生的dna的尺寸分布较短,所以我们预测由非删除的单倍型对应的片段的尺寸分布比由删除的单倍型对应的片段的尺寸分布更短。可以通过将片段的累积频率对dna片段的尺寸绘图来测定两个尺寸分布的差异。尺寸分布较短的dna群体会具有相对较多的短dna片段,因此,在尺寸范围的较短片段对应的区间中,尺寸分布较短的dna群体对应的累积频率会更快速地增加。
[0264]
图25示出了根据本发明的实施方案,针对肿瘤组织中表现出loh的区域,血浆dna的累积频率与尺寸之间的关系。x轴以碱基对为单位片段的尺寸。y轴为尺寸小于x轴上的值所对应的片段百分率。与由删除的单倍型得到的序列相比,由非删除的单倍型得到的序列具有更快速的增多且尺寸小于170bp对应的累积频率更高。这表明由非删除的单倍型得到的短dna片段是更富足的。由于由非删除的单倍型得到的肿瘤衍生的短dna的尺寸分布,上述情况与上文的预测一致。
[0265]
在一个实施方案中,可以通过在两个dna分子群体的累积频率中的差异来度量尺寸分布的差异。我们将δq定义为两个群体的累积频率的差异。δq=q
非删除的
–q删除的
,q
非删除的
表
示由非删除对应的单倍型得到的测序dna片段的累积频率;而q
删除的
表示由删除对应的单倍型得到的测序dna片段的累积频率。
[0266]
图26示出了δq与loh区域的测序血浆dna的尺寸之间的关系。根据本发明的实施方案,在尺寸为130bp时,δq达到0.2。这表明使用130bp作为用于定义短dna的截断值是最佳的,以用于上文所述的等式中。使用这种截断值,当与得自删除的单倍型的群体相比较时,在由非删除的单倍型的群体中,短dna分子量高出20%之多。接着,将该百分率差异(或相似的衍生值)与由未患有癌症的个体推导得到的阈值相比。
[0267]
就具有染色体扩增的区域而言,在肿瘤组织中,一个单倍型被复制(扩增的单倍型)。由于由这种扩增的单倍型得到的额外量的肿瘤衍生的短dna分子被释放到血浆中,则由扩增的单倍型得到的片段的尺寸分布比由非扩增的单倍型得到的片段的尺寸分布更短。与loh方案相似,可以针对dna片段的尺寸绘制片段的累积频率来测定尺寸分布的差异。具有更短的尺寸分布的dna的群体具有更多的短dna,因此在尺寸范围短片段所对应的区间中,其累积频率会更快速地增加。
[0268]
图27示出了根据本发明的实施方案,血浆dna的累积频率对肿瘤组织中具有染色体复制的区域所对应的的dna片段尺寸的关系。与由非扩增的单倍型得到的序列相比,由扩增的单倍型得到的序列具有更快速的增多而且尺寸小于170bp对应的累积频率更高的。这表明,由扩增的单倍型得到的短dna片段量更多。由于大量的肿瘤衍生的短dna是由扩增的单倍型衍生的,所以上述情况与下文所示的预计是一致的。
[0269]
与loh方案相似,可以通过两个群体的dna分子的累积频率的差异来定量分析尺寸分布的差异。我们将δq定义为两个群体的累积频率的差异。δq=q
扩增的
–q非扩增的
,q
扩增的
表示由扩增的单倍型得到的测序dna片段的累积频率;而q
非扩增的
表示由非扩增的单倍型得到的测序dna片段的累积频率。
[0270]
图28示出了根据本发明的实施方案,针对扩增的区域,δq与测序血浆dna的尺寸之间的关系。根据本发明的实施方案,在尺寸为126bp时,δq达到0.08。这表明使用126bp作为用于定义短dna的截断值,在与由非扩增的单倍型得到的群体相比时,在由扩增的单倍型得到的群体中,短dna分子量更多,即高出8%之多。
[0271]
d.其他技术
[0272]
在其他的实施方案中,可以使用序列特异性的技术。例如,可以设计寡核苷酸从而与特定区域的片段杂交。然后,按照与测序标签计数相似的方式,计数寡核苷酸。该方法可以用于检测展现特定畸变的癌症。
[0273]
viii.计算机系统
[0274]
本文提及的任何计算机系统可以使用任何合适数量的亚系统。此类亚系统的实例示于图9的计算机仪器900中。在一些实施方案中,计算机系统包括单一的计算机仪器,其中亚系统可以为计算机仪器的部件。在其他的实施方案中,计算机系统可以包括多个计算机仪器(每个为亚系统)以及内部部件。
[0275]
图29所示的亚系统通过系统总线2975相互连接。示出与显示适配器2982偶联的其他亚系统,例如打印机2974、键盘2978、硬盘2979、监控器2976等。外部和输入/输出(i/o)装置(其与i/o控制器2971偶联)可以通过本领域已知的任意一种手段(串行端口2977)与计算机系统连接。例如,串行端口2977或外部界面2981(例如ethernet、wi-fi等)可以用于将计
算机系统2900与诸如internet、鼠标输入装置或扫描器之类的广域网路连接。通过系统总线2975的相互连接允许中央处理器2973与各个亚系统连通,并由中央处理器2973控制系统储存器2972或硬盘2979对指令执行,以及控制亚系统之间的信息交换。系统储存器2972和/或硬盘2979可以表现为计算机可读介质。本文提及的任何值都可以由一个部件输出至另一个部件,并可以输出给用户。
[0276]
计算机系统可以包括例如通过外部界面2981或通过内部界面连接在一起的多个相同的部件或亚系统。在一些实施方案中,计算机系统、亚系统或仪器可以在网络上连通。在这种情况下,一个计算机可以为认为是一个客户端及另一个计算机的服务器,其中它们均可以为同一计算机系统的部件。客户端和服务器均可以包括多个系统、亚系统或部件。
[0277]
应该这样理解,本发明的任意实施方案均可以使用硬件和/或使用计算机软件以模块或集成的方式以控制逻辑的形式来实现。基于本文所提供的公开的发明和指导说明,本领域的普通技术人员将了解并领会使用硬件以及硬件和软件的组合来实现本发明的实施方案的其他方式和/或方法。
[0278]
本技术中所描述的任意软件部件或功能都可以作为使用任何合适的计算机语言(例如java,c++或perl)通过处理器执行的软件编码(基于传统或面向对象的技术)来实施。软件编码可以在用于储存和/或传输的计算机可读介质上以一系列指令或命令的形式储存,合适的介质包括随机储存器(ram)、只读储存器(rom)、磁介质(例如硬盘机或软盘)、或光学介质(例如光盘(cd)或dvd(数字通用光盘))、闪存等。计算机可读介质可以为此类储存或传输装置的任意组合。
[0279]
此外,此类程序还可以使用适用于通过有线、光学和/或无线网络(遵守多种协议,包括internet)传输的载波信号来编码和传输。由此,可以使用由此类程序编码的数字信号来创建根据本发明的实施方案的计算机可读介质。使用程序编码所编码的计算接可读介质可以嵌入到兼容的装置,或者由其他装置分开提供(例如通过internet下载)。任意此类的计算机可读介质可以存在单一的计算机程序产品(例如硬盘机、cd或整个计算机系统)上或内,并且可以在系统或网络内的不同计算机程序产品上或该产品内存在。计算机系统可以包括监控器、打印机或用于向用户提供本文所提及的任意结果的其他合适的显示器。
[0280]
本文所述的任何方法可以使用计算机系统(包括处理器)全部或部分实现,所述的系统可以被配置成实现所述的步骤。因此,多个实施方案可以针对于计算机系统而言,该系统被构造成潜在地使用实施各个步骤或各组步骤的不同部件来实施本文所述的任意方法的步骤。尽管本文所述的方法以编号的步骤呈现,但是这些方法的步骤可以同时或以不同的次序来实施。此外,这些步骤的一部分可以与其他方法的其他步骤的一部分一起使用。此外,步骤的全部或一部分可以是任选的。此外,任意方法的任意步骤可以使用模块、电路或用于实施这些步骤的其他手段来实施。
[0281]
在不脱离本发明的实质和范围的条件下,特定实施方案的具体详情可以以任何合适的方式来结合。但是,本发明的其他实施方案可以针对各单一的方面或这些单一方面的特定组合有关的特定实施方案。
[0282]
为了进行说明和描述本发明,上述描述已经呈现了本发明的示例性实施方案。无意于穷举或将本发明完全限定于示例所述的形式,并且根据上文的发明说明,许多修改和改变的版本也是可行的。选择并描述多个实施方案,以便最好地说明本发明的原理及其实
践应用,由此使本领域的技术人员能够在各种实施方案以及各种修改(其适用于所考虑的特定用途)中最好地利用本发明。
[0283]
除非特异作出相反的说明,描述“一个”(“a”或“an”)、或“这”(“the”)意指“一个或多个”。就所有的目的而言,上文提及的所有专利、专利申请、公开发明和描述在此以引用方式作为整体并入本文。这些文件均未确认为现有技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1