一种对多物种进行多核苷酸变异鉴定和注释的方法
1.本发明属于生物技术领域,具体的说就是一种对多物种进行多核苷酸变异鉴定和注释的方法
背景技术:
2.近年来,很多国家先后投入大量资金启动精准医疗计划,人类已逐渐走进精准医疗时代。因此,解析不同个体遗传差异,是精准医疗实行的一个重要前提。随着技术发展,获得遗传信息的时间和成本大幅度降低,人类疾病研究中鉴定到了大量的多核苷酸变异(multi-nucleotide variants,mnvs)。mnv指个体中同一个单倍型上同时存在两个或两个以上的核苷酸变异,会导致蛋白质序列的改变,相较于单核苷酸变异(同一个单倍型上单点的核苷酸变异)可能具有更高的致害性。目前,mnv的鉴定还仅仅只涉及到双点mnv的鉴定,而且传统的遗传变异注释工具(数据库和软件)通常无法对大于双点的mnv进行注释。
技术实现要素:
3.本发明是为了解决上述现有技术存在的不足之处,提供一种对多物种进行多核苷酸变异鉴定和注释的方法,不仅可以获得大于双点的mnv(至多可以鉴定到7个位点mnv),同时也对这些mnv进行功能的注释,方便科研工作者根据科研需求对鉴定和注释后的mnv数据进行筛选、探讨和研究。
4.本发明为了达到上述发明目的,采用如下技术方案:
5.一种对多物种进行多核苷酸变异鉴定和注释的方法,包括以下步骤:
6.步骤1、根据已经定向好的数据集vcf,获得所有双点mnv为数据集twopointmnv;
7.步骤2、将数据集twopointmnv中所有鉴定到的双点mnv拆成单点作为数据集snv,并根据数据集snv从数据集vcf中获取单点的行信息作为数据集snvinfovcf;
8.步骤3、将数据集snv复制6份,分别记作第六副本数据集snv7~第一副本数据集snv2;
9.步骤4、鉴定7位点至2位点mnv,获得鉴定后的mnv。
10.如上所述的步骤4包括以下步骤:
11.步骤4.1、初始定义副本数据集序号n为6,初始定义位点组合序号m为n+1,初始定义组合合计次数号k为n+1;
12.步骤4.2、选用第n副本数据集snv(n+1),以10bp宽度的窗口扫描第n副本数据集snv(n+1)、枚举所有可能的m位点组合为数据集snv(n+1)sub;
13.步骤4.3、提取数据集snv(n+1)sub中第一个m位点组合;
14.步骤4.4、将该m位点组合拆成单点再从数据集snvinfovcf中获取单点的行信息并进行统计:分别统计m位点组合中合计为m~2的次数;
15.步骤4.5、对统计结果进行判断:如果m位点组合合计为k的次数等于0,那么该m位点组合不是mnv,从数据集snv(n+1)sub中删除该m位点组合,提取数据集snv(n+1)sub中下
一个m位点组合并回到步骤4.4;如果m位点组合合计为k的次数大于0,那么该m位点组合为mnv,保留该m位点组合并追加mnv距离、检测到该mnv的单倍型数量和频率三个信息作为鉴定后的mnv;
16.步骤4.6、k自减1;
17.步骤4.7、如果m位点组合合计为k的次数大于0,提取数据集snv(n+1)sub中下一个m位点组合并回到步骤4.4;
18.如果m位点组合合计为k的次数等于0,则删除第(k-1)副本snv(k)中存在于m位点组合中的所有单点;
19.步骤4.8、k自减1,返回步骤4.7,直至k自减后等于1,进入步骤4.9;
20.步骤4.9、副本数据集序号n自减1,定义位点组合序号m为n+1,定义组合合计次数号k为n+1,返回步骤4.2,直至副本数据集序号n自减1后等于0。
21.一种对多物种进行多核苷酸变异鉴定和注释的方法,还包括以下步骤:
22.步骤5、将鉴定后的mnv作为数据集allmnv;
23.步骤6、将数据集allmnv与预先内置好的多核苷酸变异注释信息数据库进行匹配,输出基于基因注释的已知数据集mnvgeneknow和未知数据集mnvgeneunknow、基于非编码区注释的已知数据集mnvnonknow和未知数据集mnvnonunknow、以及基于调控区注释的已知数据集mnvregknow和未知数据集mnvregunknow。
24.一种对多物种进行多核苷酸变异鉴定和注释的方法,还包括以下步骤:
25.步骤7、基于基因注释,具体包括:
26.步骤7.1、根据选择的物种的参考基因组注释文件和cds序列文件进行数据集geneanno构建,对数据集geneanno中所有转录本进行分组,获得分组区段数据集generange;
27.步骤7.2、将未知数据集mnvgeneunknow的mnv拆成单点为数据集singlepoint;
28.步骤7.3、提取数据集singlepoint中的第一个点;
29.步骤7.4、将数据集singlepoint中提取的点在分组区段数据集generange进行定位,确定这个点落在分组区段数据集generange的分组区段从而获得落在该分组区段的所有转录本;
30.步骤7.5、对步骤7.4获得的分组区段的所有转录本进行遍历,确定步骤7.4中的点具体落在的转录本以及转录本的区段,将转录本名、对应的基因名、对应的基因常用名、落在转录本上的具体区段追加到数据集singlepoint中提取的点后;
31.步骤7.6、提取数据集singlepoint中的下一个点,返回步骤7.4,直至遍历数据集singlepoint中所有点;
32.步骤7.7、提取未知数据集mnvgeneunknow中的第一个mnv,在数据集singlepoint中提取mnv中单点的信息并整合到对应的未知数据集mnvgeneunknow中提取的mnv后,重复本步骤直至遍历提取未知数据集mnvgeneunknow中所有mnv;
33.步骤7.8、将上述的未知数据集mnvgeneunknow和已知数据集mnvgeneknow合并输出为数据集mnvgene。
34.一种对多物种进行多核苷酸变异鉴定和注释的方法,还包括以下步骤:
35.步骤8、基于非编码区注释,具体包括:
36.步骤8.1、对选择的物种,整合非编码区注释和第三方的非编码区注释,获得数据集noncodinganno,将数据集noncodinganno中所有非编码区段进行分组,获得区段数据集noncodingrange:
37.步骤8.2、提取未知数据集mnvnonunknow中的第一个mnv;
38.步骤8.3、将该mnv在区段数据集noncodingrange中进行定位,确定mnv落在区段数据集noncodingrange的分组区段从而获得落在该分组区段的所有非编码区段;
39.步骤8.4、对步骤8.3中落在分组区段的所有非编码区段进行遍历,从而确定步骤8.3中的mnv具体落在的非编码区段,把非编码区段类型、名称和来源追加到数据集mnvnonunknow中提取的点后;
40.步骤8.5、提取未知数据集mnvnonunknow中的下一个mnv,返回步骤8.3,直至遍历未知数据集mnvnonunknow中所有的mnv;
41.步骤8.6、将上述的未知数据集mnvnonunknow和已知数据集mnvnonknow合并输出为数据集mnvnon。
42.一种对多物种进行多核苷酸变异鉴定和注释的方法,还包括以下步骤:
43.步骤9、基于调控区注释,具体包括以下步骤:
44.步骤9.1、根据第三方的调控区注释,对选择的物种构建数据集regulatoranno,将数据集regulatoranno中所有调控区段进行分组,获得区段数据集regulatorrange;
45.步骤9.2、提取未知数据集mnvregunknow中的第一个mnv;
46.步骤9.3、将该mnv在区段数据集regulatorrange中进行定位,确定mnv落在区段数据集regulatorrange的分组区段从而获得落在该分组区段的所有调控区段;
47.步骤9.4、对步骤9.3中落在分组区段的所有调控区段进行遍历,从而确定步骤9.3中的mnv具体落在的调控区段,把调控区段类型、名称和来源追加到数据集mnvregunknow中提取的点后;
48.步骤9.5、提取未知数据集mnvregunknow中的下一个mnv,返回步骤9.3,直至遍历未知数据集mnvregunknow中所有的mnv;
49.步骤9.6、将未知数据集mnvregunknow和已知数据集mnvregknow合并输出为数据集mnvreg。
50.一种对多物种进行多核苷酸变异鉴定和注释的方法,还包括以下步骤:
51.步骤10、将数据集mnvgene、数据集mnvnon和数据集mnvreg合并输出为数据集mnvanno。
52.本发明相对于现有技术,具有以下有益效果:
53.1、本发明方法拓展了传统的mnv鉴定方法,支持大于双点的mnv的鉴定,不仅增加鉴定到的mnv数量,而且也过滤掉原先被错误鉴定的mnv;2、传统的遗传变异注释工具不是为mnv开发的,无法对其进行有效注释,本发明专门为mnv进行注释,不仅避免了这些错误,而且也减少了大量的时间,为科研工作者提供一个研究mnv便利高效的工具。
附图说明
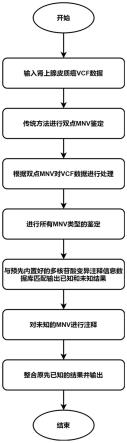
54.图1为本发明的流程图:
具体实施方式
55.为了便于本领域普通技术人员理解和实施本发明,下面结合实施例对本发明作进一步的详细描述,应当理解,此处所描述的实施示例仅用于说明和解释本发明,并不用于限定本发明。
56.本实施例中,一种对多物种进行多核苷酸变异鉴定和注释的方法,从物种定向后的标准变异格式数据(the variant call format,vcf)中鉴定mnv并进行注释。具体的讲,如图1所示,按照如下步骤进行:
57.步骤1、采用已经定向好的肾上腺皮质癌标准变异格式数据(以下简称为数据集vcf)。先利用传统的多核苷酸变异鉴定工具(identify_mnv,https://github.com/macarthur-lab/gnomad_mnv)获取所有双点mnv为数据集twopointmnv。
58.步骤2、根据数据集twopointmnv的结果,将所有鉴定到的双点mnv拆成单点作为数据集snv,并根据数据集snv从数据集vcf中获取单点的行信息作为数据集snvinfovcf。
59.步骤3、将数据集snv复制6份,分别记作第六副本数据集snv7~第一副本数据集snv2。
60.步骤4、鉴定7位点至2位点mnv,获得鉴定后的mnv,具体包括以下步骤:
61.步骤4.1、初始定义副本数据集序号n为6,初始定义位点组合序号m为n+1,初始定义组合合计次数号k为n+1;
62.步骤4.2、选用第n副本数据集snv(n+1),以10bp宽度的窗口扫描第n副本数据集snv(n+1)、枚举所有可能的m位点组合为数据集snv(n+1)sub;
63.步骤4.3、提取数据集snv(n+1)sub中第一个m位点组合;
64.步骤4.4、将该m位点组合拆成单点再从数据集snvinfovcf中获取单点的行信息并进行统计:分别统计m位点组合中合计为m~2的次数(例如,m为7时,一个样本的一条单倍型上7个点都是1,则合计为7,出现的次数记为1,一个样本的一条单倍型上6个点都是1,则合计为6,其出现的次数记为1,依次类推,一个样本的一条单倍型上2个点都是1,则合计为2,其出现的次数记为1);
65.步骤4.5、对统计结果进行判断:如果m位点组合合计为k的次数等于0,那么该m位点组合不是mnv,从数据集snv(n+1)sub中删除该m位点组合,提取数据集snv(n+1)sub中下一个m位点组合并回到步骤4.4;如果m位点组合合计为k的次数大于0,那么该m位点组合为mnv,保留该m位点组合并追加3个信息(mnv距离、检测到该mnv的单倍型数量和频率)作为鉴定后的mnv;
66.步骤4.6、k自减1;
67.步骤4.7、如果m位点组合合计为k的次数大于0,提取数据集snv(n+1)sub中下一个m位点组合并回到步骤4.4;
68.如果m位点组合合计为k的次数等于0,则删除第(k-1)副本snv(k)中存在于m位点组合中的所有单点;
69.步骤4.8、k自减1,返回步骤4.7,直至k自减后等于1,进入步骤4.9;
70.步骤4.9、副本数据集序号n自减1,定义位点组合序号m为n+1,定义组合合计次数号k为n+1,返回步骤4.2,直至副本数据集序号n自减1后等于0,则进入下一步。
71.对步骤4进行举例说明:
72.(a)鉴定7位点mnv
73.选用第六副本数据集snv7。以10bp宽度的窗口扫描第六副本数据集snv7、枚举所有可能的7位点组合为数据集snv7sub。
74.提取数据集snv7sub中第一个7位点组合,将该7位点组合拆成单点再从数据集snvinfovcf中获取单点的行信息并进行统计:统计7位点组合中合计为7的次数(一个样本的一条单倍型上7个点都是1,则合计为7,出现的次数记为1),合计为6的次数(一个样本的一条单倍型上6个点都是1,则合计为6,其出现的次数记为1),以此类推,一直统计到合计为2的次数(一个样本的一条单倍型上2个点都是1,则合计为2,其出现的次数记为1)。
75.对统计结果进行判断:如果7位点组合合计为7的次数等于0,那么该7位点组合不是mnv,从数据集snv7sub中删除该7位点组合,提取数据集snv7sub中下一个7位点组合并回到步骤4.4;如果7位点组合合计为7的次数大于0,那么该7位点组合为mnv,保留该7位点组合并追加3个信息(mnv距离、检测到该mnv的单倍型数量和频率)作为鉴定后的mnv。
76.判断7位点组合合计为6的次数,如果大于0,提取数据集snv7sub中下一个7位点组合并回到步骤4.4;如果合计为6的次数等于0,则删除第五副本snv6中存在于7位点组合中的所有单点,并判断7位点组合合计为5的次数。如果7位点组合合计为5的次数大于0,提取数据集snv7sub中下一个7位点组合并回到步骤4.4;如果合计为5的次数等于0,则删除第四副本snv5中存在于7位点组合中的所有单点,并判断7位点组合合计为4的次数,以此类推。
77.(b)鉴定6位点mnv
78.选用第五副本数据集snv6。以10bp宽度的窗口扫描第五副本数据集snv6、枚举所有可能的6位点组合为数据集snv6sub。
79.提取数据集snv6sub中第一个6位点组合,将该6位点组合拆成单点再从数据集snvinfovcf中获取单点的行信息并进行统计:统计6位点组合中合计为6的次数(一个样本的一条单倍型上6个点都是1,则合计为6,出现的次数记为1),合计为5的次数(一个样本的一条单倍型上5个点都是1,则合计为5,其出现的次数记为1),以此类推,一直统计到合计为2的次数(一个样本的一条单倍型上2个点都是1,则合计为2,其出现的次数记为1)。
80.对统计结果进行判断:如果6位点组合合计为6的次数等于0,那么该6位点组合不是mnv,从数据集snv6sub中删除该组合,提取数据集snv6sub中下一个6位点组合并回到步骤4.4;如果6位点组合合计为6的次数大于0,那么该6位点组合为mnv,保留该6位点组合并追加3个信息(mnv距离、检测到该mnv的单倍型数量和频率)作为鉴定后的mnv。
81.判断6位点组合合计为5的次数,如果大于0,提取数据集snv6sub中下一个6位点组合并回到步骤4.4;如果合计为5的次数等于0,则删除第四副本snv5中存在于6位点组合中的所有单点,并判断6位点组合合计为4的次数。如果6位点组合合计为4的次数大于0,提取数据集snv6sub中下一个6位点组合并回到步骤4.4;如果合计为4的次数等于0,则删除第三副本snv4中存在于6位点组合中的所有单点,并判断6位点组合合计为3的次数,以此类推。
82.步骤5、经过上述循环后将输出所有类型的鉴定后的mnv作为数据集allmnv。下面开始对这些mnv进行注释,注释包括3种类型:基于基因注释(针对的是蛋白编码基因)、基于非编码区注释和基于调控区注释。
83.步骤6、将数据集allmnv与预先内置好的多核苷酸变异注释信息数据库(在之前的研究中已经被鉴定和注释好的mnv)进行匹配,输出6个结果分别是:基于基因注释的已知数
据集mnvgeneknow和未知数据集mnvgeneunknow、基于非编码区注释的已知数据集mnvnonknow和未知数据集mnvnonunknow、基于调控区注释的已知数据集mnvregknow和未知数据集mnvregunknow。
84.步骤7、基于基因注释。
85.步骤7.1、根据选择的物种的参考基因组注释文件和cds序列文件进行数据集geneanno构建,对数据集geneanno中所有转录本进行分组,获得分组区段数据集generange:
86.1)获取物种的参考基因组注释文件和cds序列文件;
87.2)处理上述信息获得数据集geneanno,数据集geneanno每行代表一个转录本,列信息由两部分组成:基础信息(转录本名、染色体、链、对应的基因名、对应的基因常用名、序列信息、转录本起始位点、转录本终止位点、cds起始位点、cds终止位点、exon数量、exon起始位点、exon终止位点)和补充信息(基因间区段、转录本上游区段、转录本下游区段、转录本5’utr区段、转录本3’utr区段,转录本exon区段、转录本splicing区段、转录本intron区段);
88.3)根据数据集geneanno中每个转录本的起始位点和终止位点,将数据集geneanno中所有转录本进行分组,每个组有一个分组区段号(该组内所有转录本起始位点的最小值-该组内所有转录本终止位点的最大值,如1-100),确保组与组之间的分组区段号不重叠(如分组区段号1-100与分组区段号200-300),最终构建分组区段数据集generange。分组区段数据集generange每行表示一个分组区段,列信息包含2列:分组区段号和落在该分组区段的所有转录本(每一个转录本包含:基础信息和补充信息,不同转录本用;分隔)。
89.步骤7.2、将未知数据集mnvgeneunknow的mnv拆成单点为数据集singlepoint。
90.步骤7.3、提取数据集singlepoint中的第一个点。
91.步骤7.4、将数据集singlepoint中提取的点在分组区段数据集generange进行定位,确定这个点落在分组区段数据集generange的哪个分组区段从而获得落在该分组区段的所有转录本。
92.步骤7.5、对步骤7.4中落在该分组区段的所有转录本进行遍历,从而确定步骤7.4中的点具体落在哪个转录本、转录本的哪个区段(基因间区段、转录本上游区段、转录本下游区段、转录本5’utr区段、转录本3’utr区段,转录本exon区段、转录本splicing区段、转录本intron区段),把这部分信息(转录本名、对应的基因名、对应的基因常用名、落在该转录本上的具体区段)追加到数据集singlepoint中提取的点后。
93.步骤7.6、提取数据集singlepoint中的下一个点,返回步骤7.4,直至遍历数据集singlepoint中所有点。
94.步骤7.7、提取未知数据集mnvgeneunknow中的第一个mnv,在数据集singlepoint中提取mnv中单点的信息并整合到对应的未知数据集mnvgeneunknow中提取的mnv后,同时计算这个mnv是否会造成氨基酸序列的改变、序列改变的类型和危险分类,并整合到对应的未知数据集mnvgeneunknow中提取的mnv后。重复本步骤直至遍历提取未知数据集mnvgeneunknow中所有mnv。
95.步骤7.8、将上述的未知数据集mnvgeneunknow和已知数据集mnvgeneknow合并输出为数据集mnvgene;
96.步骤8、基于非编码区注释。
97.步骤8.1、对选择的物种,整合非编码区注释和第三方的非编码区注释,获得数据集noncodinganno,将数据集noncodinganno中所有非编码区段进行分组,获得区段数据集noncodingrange:
98.1)获取物种的参考基因组注释文件并从参考基因组注释文件中获取非编码区注释;
99.2)获取其他第三方的非编码区注释(包括mirbase、noncode、circbase和gencode等);
100.3)整合非编码区注释和第三方的非编码区注释,获得数据集noncodinganno,数据集noncodinganno的每行代表一个非编码区段、列信息包含8列:非编码区段类型、染色体、起始位点、终止位点、链、名称、来源和其他信息如疾病信息;
101.4)根据数据集noncodinganno中每个非编码区段的起始位点和终止位点,将数据集noncodinganno中所有非编码区段进行分组,每个组有一个分组区段号(该组内所有非编码区段起始位点的最小值-该组内所有非编码区段终止位点的最大值,如1-100),确保组与组之间的分组区段号不重叠(如分组区段号1-100与分组区段号200-300),最终构建区段数据集noncodingrange。区段数据集noncodingrange每行表示一个分组区段,列信息包含2列:分组区段号和落在该分组区段的所有非编码区段(每一个非编码区段包含:非编码区段类型、染色体、起始位点、终止位点、链、名称、来源和其他信息,不同非编码区段用“;”分隔)。
102.步骤8.2、提取未知数据集mnvnonunknow中的第一个mnv。
103.步骤8.3、将该mnv在区段数据集noncodingrange中进行定位,确定这个mnv落在区段数据集noncodingrange的哪个分组区段从而获得落在该分组区段的所有非编码区段。
104.步骤8.4、对步骤8.3中落在该分组区段的所有非编码区段进行遍历,从而确定步骤8.3中的mnv具体落在哪个非编码区段,把这部分信息(非编码区段类型、名称、来源和其他信息)追加到数据集mnvnonunknow中提取的点后。
105.步骤8.5、提取未知数据集mnvnonunknow中的下一个mnv,返回步骤8.3,直至遍历未知数据集mnvnonunknow中所有的mnv。
106.步骤8.6、将上述的未知数据集mnvnonunknow和已知数据集mnvnonknow合并输出为数据集mnvnon。
107.步骤9、基于调控区注释。
108.步骤9.1、根据第三方的调控区注释,对选择的物种构建数据集regulatoranno,将数据集regulatoranno中所有调控区段进行分组,获得区段数据集regulatorrange:
109.1)获取第三方的调控区注释(包括fantom5,atacdb,ucsc等)并整合获得数据集regulatoranno,每行代表一个调控区段、列信息包含8列:调控区段类型、染色体、起始位点、终止位点、链、名称、来源和其他信息如疾病信息;
110.2)根据数据集regulatoranno中每个调控区段的起始位点和终止位点,将数据集regulatoranno中所有调控区段进行分组,每个组有一个分组区段号(该组内所有调控区段起始位点的最小值-该组内所有调控区段终止位点的最大值,如1-100),确保组与组之间的分组区段号不重叠(如分组区段号1-100与分组区段号200-300),最终构建区段数据集
regulatorrange。区段数据集regulatorrange每行表示一个分组区段,列信息包含2列:分组区段号和落在该分组区段的所有调控区段(每一个调控区段包含:调控区段类型、染色体、起始位点、终止位点、链、名称、来源和其他信息,不同调控区段用“;”分隔)。
111.步骤9.2、提取未知数据集mnvregunknow中的第一个mnv。
112.步骤9.3、将该mnv在区段数据集regulatorrange中进行定位,确定这个mnv落在区段数据集regulatorrange的哪个分组区段从而获得落在该分组区段的所有调控区段。
113.步骤9.4、对步骤9.3中落在该分组区段的所有调控区段进行遍历,从而确定步骤9.3中的mnv具体落在哪个调控区段,把这部分信息(调控区段类型、名称、来源和其他信息)追加到数据集mnvregunknow中提取的点后。
114.步骤9.5、提取未知数据集mnvregunknow中的下一个mnv,返回步骤9.3,直至遍历未知数据集mnvregunknow中所有的mnv。
115.步骤9.6、将上述的未知数据集mnvregunknow和已知数据集mnvregknow合并输出为数据集mnvreg。
116.步骤10、将数据集mnvgene、数据集mnvnon和数据集mnvreg合并输出为数据集mnvanno。
117.准备的数据集
118.本发明使用肾上腺皮质癌vcf数据,该数据是真实数据集。在该数据集中,包含22492行(单核苷酸位点数量)和9+77列(基础信息+人类肾上腺皮质癌样本),使用全部数据来验证其鉴定到的mnv数量与传统多核苷酸变异鉴定方法的结果比较。实验结果如表1所示。
119.表1本发明的多核苷酸变异的鉴定方法较传统鉴定方法的鉴定结果比对表
120.方法双点mnv>2位点mnv总数改进的多核苷酸变异鉴定43717454传统的多核苷酸变异鉴定4940494
121.从表1可以看出在肾上腺皮质癌vcf数据中,本发明的多核苷酸变异的鉴定方法较传统鉴定方法鉴定出了额外的其他类型的mnv共17个,以及删除了原先被错误鉴定为双点mnv共57个。
122.本发明还同时对所有得到的mnv进行注释。
123.需要指出的是,本发明中所描述的具体实施例仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例作各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或超越所附权利要求书所定义的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1