碳纤维前体用处理剂的水性液和碳纤维前体的制作方法
1.本发明涉及能够提高经时稳定性和碳纤维的强度的碳纤维前体用处理剂的水性液、以及赋予该碳纤维前体用处理剂的水性液而得到的碳纤维前体。
背景技术:
2.碳纤维一般作为例如与环氧树脂等基体树脂组合而成的碳纤维复合材料被广泛用于建材、传输设备等各领域中。碳纤维通常作为碳纤维前体经过例如对丙烯酸纤维进行纺丝的工序、将纤维拉伸的工序、耐火处理工序、以及碳化处理工序来制造。对于碳纤维前体,为了提高碳纤维的制造工序中的纤维的集束性或者抑制纤维间的胶着或熔接,使用了碳纤维前体用处理剂。
3.以往已知有专利文献1~3中所公开的碳纤维前体用处理剂。专利文献1中公开了一种氨基改性硅油剂组合物,其含有包含具有特定粘度的氨基改性聚硅氧烷的硅油剂以及包含非离子表面活性剂的乳化剂等。专利文献2中公开了一种合成纤维处理油剂,其含有具有特定化学式的多环芳香族系非离子乳化剂、氨基改性硅酮等。专利文献3中公开了一种硅油组合物,其含有硅酮、由烷基链和聚氧亚烷基链形成的非离子表面活性剂。现有技术文献专利文献
4.专利文献1:日本特许第3459305号公报专利文献2:日本特许第4311246号公报专利文献3:日本特开2005
‑
298689号公报
技术实现要素:
发明所要解决的课题
5.但是,现有的碳纤维前体用处理剂在以水性液的状态保存时,具有经时稳定性降低、最终得到的碳纤维的强度降低的问题。
6.本发明所要解决的课题在于提供能够提高经时稳定性和碳纤维的强度的碳纤维前体用处理剂的水性液以及碳纤维前体。用于解决课题的手段
7.于是,本发明人为了解决上述课题进行了研究,结果发现,除了氨基改性硅酮以外还合用了具有特定分子量分布的非离子表面活性剂的碳纤维前体用处理剂的水性液是恰好合适的。
8.作为用于解决上述课题的本发明的一个方式的碳纤维前体用处理剂的水性液的特征在于,其包含碳纤维前体用处理剂以及水,该碳纤维前体用处理剂含有氨基改性硅酮和分子量分布(mw/mn)为1.05~1.50的非离子表面活性剂。
9.上述碳纤维前体用处理剂的水性液中,上述非离子表面活性剂可以由下述化1所表示。
10.[化1](化1中,r1:碳原子数8~18的直链烃基或碳原子数8~18的支链烃基;ao:碳原子数2~3的氧化亚烷基;r2:氢原子或碳原子数1~4的烃基;n:1~60的整数。)上述碳纤维前体用处理剂的水性液中,上述非离子表面活性剂可以包含上述化1中的r1的碳原子数不同的2种以上的非离子表面活性剂。
[0011]
上述碳纤维前体用处理剂的水性液中,上述氨基改性硅酮在25℃的运动粘度可以为50~4000mm2/s。
[0012]
上述碳纤维前体用处理剂的水性液中,将上述氨基改性硅酮、上述水以及上述非离子表面活性剂的合计含有比例设为100质量份时,上述氨基改性硅酮和上述非离子表面活性剂的合计含有比例可以为20~50质量份,并且可以以50~80质量份的比例含有上述水。
[0013]
上述碳纤维前体用处理剂的水性液中,上述氨基改性硅酮的含量相对于上述非离子表面活性剂的含量的质量比可以为上述氨基改性硅酮/上述非离子表面活性剂=95/5~75/25。
[0014]
作为本发明的另一方式的碳纤维前体的特征在于,其附着有上述碳纤维前体用处理剂。发明的效果
[0015]
根据本发明,能够提高经时稳定性和碳纤维的强度。
具体实施方式
[0016]
(第1实施方式)下面对具体实现本发明的碳纤维前体用处理剂的水性液(以下简称为水性液)的第1实施方式进行说明。本实施方式的水性液中,除了氨基改性硅酮以外,还含有具有特定分子量分布的非离子表面活性剂作为必要成分。
[0017]
氨基改性硅酮具有由(
‑
si
‑
o
‑
)重复单元形成的聚硅氧烷骨架,其硅原子的烷基侧链的一部分被氨基改性基团进行了改性。氨基改性基团可以与作为主链的硅酮的侧链键合,也可以与末端键合,还可以与这两者键合。作为氨基改性基团,例如可以举出氨基、具有氨基的有机基团等。作为具有氨基的有机基团,可例示出下述化2。
[0018]
[化2]
‑
r1‑
(nh
‑
r2)
z
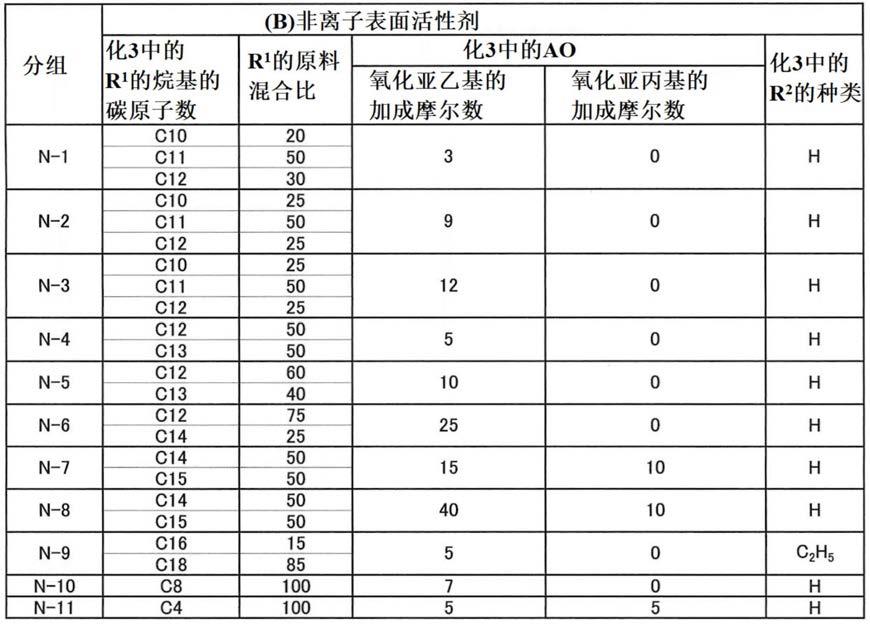
‑
nh2(化2中,r1和r2为碳原子数2~4的亚烷基,分别可以相同也可以不同。z为0或1的整数。)作为化2的具有氨基改性基团的氨基改性硅酮的具体例,例如可以举出二甲基硅
氧烷
‑
甲基(氨基丙基)硅氧烷共聚物(氨基丙基聚二甲基硅氧烷)、氨基乙基氨基丙基甲基硅氧烷
‑
二甲基硅氧烷共聚物(氨端聚二甲基硅氧烷)等。
[0019]
氨基改性硅酮在25℃的运动粘度的下限没有特别限制,优选为50mm2/s以上。运动粘度的下限为50mm2/s以上的情况下,可进一步提高碳纤维的强度。氨基改性硅酮在25℃的运动粘度的上限没有特别限制,优选为4000mm2/s以下。运动粘度的上限为4000mm2/s以下的情况下,可进一步提高水性液的经时稳定性。需要说明的是,氨基改性硅酮使用2种以上的情况下的运动粘度应用所使用的2种以上的氨基改性硅酮的混合物的实际测定值。
[0020]
非离子表面活性剂的分子量分布(mw/mn)具有1.05~1.50的范围。非离子表面活性剂的分子量分布中,将混配在水性液中的非离子表面活性剂的混合物作为试验试样通过gpc法来求出。混配在水性液中的非离子表面活性剂使用非单分散而具有特定分子量分布的宽分布状态的试剂,由此可提高水性液的经时稳定性。
[0021]
关于非离子表面活性剂的种类,只要可满足上述分子量分布条件就没有特别限制,例如可以举出在醇类或羧酸类上加成环氧烷而成的化合物、羧酸类与多元醇的酯化合物、在羧酸类与多元醇的酯化合物上加成环氧烷而成的醚
‑
酯化合物等。
[0022]
关于作为非离子表面活性剂的原料使用的醇类的具体例,例如可以举出:(1)甲醇、乙醇、丙醇、丁醇、戊醇、己醇、辛醇、壬醇、癸醇、十一醇、十二醇、十三醇、十四醇、十五醇、十六醇、十七醇、十八醇、十九醇、二十醇、二十一醇、二十二醇、二十三醇、二十四醇、二十五醇、二十六醇、二十七醇、二十八醇、二十九醇、三十醇等直链烷基醇;(2)异丙醇、异丁醇、异己醇、2
‑
乙基己醇、异壬醇、异癸醇、异十三醇、异十四醇、异三十醇、异十六醇、异十七醇、异十八醇、异十九醇、异二十醇、异二十一醇、异二十二醇、异二十三醇、异二十四醇、异二十五醇、异二十六醇、异二十七醇、异二十八醇、异二十九醇、异十五醇等支链烷基醇;(3)十四碳烯醇、十六碳烯醇、十七碳烯醇、十八碳烯醇、十九碳烯醇等直链烯基醇;(4)异十六碳烯醇、异十八碳烯醇等支链烯基醇;(5)环戊醇、环己醇等环状烷基醇;(6)苯酚、苯甲醇、单苯乙烯化苯酚、二苯乙烯化苯酚、三苯乙烯化苯酚等芳香族系醇;等等。
[0023]
关于作为非离子表面活性剂的原料使用的羧酸类的具体例,例如可以举出:(7)辛酸、壬酸、癸酸、十一酸、十二酸、十三酸、十四酸、十五酸、十六酸、十七酸、十八酸、十九酸、二十酸、二十一酸、山嵛酸等直链烷基羧酸;(8)2
‑
乙基己酸、异十二酸、异十三酸、异十四酸、异十六酸、异十八酸等支链烷基羧酸;(9)十八碳烯酸、十八碳二烯酸、十八碳三烯酸等直链烯基羧酸;(10)苯甲酸等芳香族系羧酸;等等。可以使用作为一种原料的醇类或羧酸类,也可以将两种以上的醇类或羧酸类组合使用。这些之中,从使分子量分布变宽、进一步提高水性液的经时稳定性的方面出发,优选将两种以上的醇类或羧酸类组合使用。
[0024]
关于作为非离子表面活性剂的原料使用的环氧烷的具体例,例如可以举出环氧乙烷、环氧丙烷等。环氧烷的加成摩尔数可适宜地设定,优选为0.1~60摩尔、更优选为1~40摩尔、进一步优选为2~30摩尔。需要说明的是,环氧烷的加成摩尔数表示相对于所投入的原料中的醇类或羧酸类1摩尔的环氧烷的摩尔数。环氧烷的加成摩尔数为0.1摩尔以上的情况下,容易将分子量分布调整为宽分布。另一方面,环氧烷的加成摩尔数为60摩尔以下的情况下,可进一步提高碳纤维的强度。
[0025]
关于作为非离子表面活性剂的原料使用的多元醇的具体例,例如可以举出乙二醇、丙二醇、1,3
‑
丙二醇、1,2
‑
丁二醇、1,3
‑
丁二醇、1,4
‑
丁二醇、1,4
‑
丁二醇、2
‑
甲基
‑
1,2
‑
丙二醇、1,5
‑
戊二醇、1,6
‑
己二醇、2,5
‑
己二醇、2
‑
甲基
‑
2,4
‑
戊二醇、2,3
‑
二甲基
‑
2,3
‑
丁二醇、甘油、2
‑
甲基
‑2‑
羟基甲基
‑
1,3
‑
丙二醇、2
‑
乙基
‑2‑
羟基甲基
‑
1,3
‑
丙二醇、三羟甲基丙烷、去水山梨糖醇、季戊四醇、山梨糖醇等。
[0026]
可以使用作为一种原料的环氧烷或多元醇,也可以将两种以上的环氧烷或多元醇组合使用。
[0027]
为了调整非离子表面活性剂的分子量分布,可通过使用2种以上的非离子表面活性剂来得到分子量分布宽的非离子表面活性剂。为了得到分子量分布宽的非离子表面活性剂,可以举出将2种以上的非离子表面活性剂进行混合的方法。另外,可通过使用2种以上的醇类或羧酸类等的原料、变更反应条件(例如使催化剂的量比通常使用量减少)而得到分子量分布宽的非离子表面活性剂。
[0028]
这些非离子表面活性剂中,优选包含下述化3所表示的化合物。通过使用该化合物,可进一步提高水性液的经时稳定性。
[0029]
[化3](化3中,r1:碳原子数8~18的直链烃基或碳原子数8~18的支链烃基;ao:碳原子数2~3的氧化亚烷基;r2:氢原子或碳原子数1~4的烃基;n:1~60的整数。)此外,非离子表面活性剂更优选包含上述化3中的r1的碳原子数不同的2种以上的非离子表面活性剂。通过使用该非离子表面活性剂,容易将分子量分布调整为宽分布。由此可进一步提高水性液的经时稳定性。
[0030]
水性液中,上述氨基改性硅酮、上述水以及上述非离子表面活性剂的含有比例没有特别限定。水性液中,将氨基改性硅酮、水以及非离子表面活性剂的合计含有比例设为100质量份时,优选氨基改性硅酮和非离子表面活性剂的合计含有比例为20~50质量份、并且以50~80质量份的比例含有水。通过限定为该混配比例,可进一步提高水性液的经时稳定性。
[0031]
水性液中,上述氨基改性硅酮的含量相对于上述非离子表面活性剂的含量的质量比没有特别限定。氨基改性硅酮的含量相对于非离子表面活性剂的含量的质量比优选为氨基改性硅酮/非离子表面活性剂=95/5~75/25。通过限定为该混合比例,可进一步提高水性液的经时稳定性。
[0032]
(第2实施方式)接着对具体实现本发明的碳纤维前体的第2实施方式进行说明。在本实施方式的碳纤维前体上附着有第1实施方式中记载的碳纤维用处理剂。
[0033]
使用本实施方式的碳纤维前体的碳纤维的制造方法中,例如使第1实施方式的水性液附着于碳纤维前体的原料纤维而得到碳纤维前体,之后根据需要进行干燥处理,接着进行制丝工序。接下来进行:将该制丝工序中制造的碳纤维前体在200~300℃、优选230~270℃的氧化性气氛中转换成耐火纤维的耐火处理工序;以及将上述耐火纤维进一步在300
~2000℃、优选300~1300℃的惰性气氛中进行碳化的碳化处理工序。
[0034]
制丝工序是对于使第1实施方式的水性液附着于碳纤维前体的原料纤维而得到的碳纤维前体进行制丝的工序,其包括附着处理工序和拉伸工序。
[0035]
附着处理工序是在对碳纤维前体的原料纤维进行纺丝后使水性液附着的工序。即,利用附着处理工序使水性液附着于碳纤维前体的原料纤维。另外,该碳纤维前体的原料纤维在刚纺丝后即被拉伸,但将附着处理工序后的高倍率拉伸特别称为“拉伸工序”。拉伸工序可以为使用高温水蒸气的湿热拉伸法,也可以为使用热辊的干热拉伸法。
[0036]
碳纤维前体的原料纤维例如可以举出丙烯酸纤维等。丙烯酸纤维优选由以聚丙烯腈(该聚丙烯腈是使至少90摩尔%以上的丙烯腈与10摩尔%以下的耐火促进成分进行共聚而得到的)作为主成分的纤维构成。作为耐火促进成分,可以适当地使用例如对于丙烯腈具有共聚性的含乙烯基化合物。关于碳纤维前体的单纤维细度没有特别限定,从性能和制造成本的平衡的方面出发,优选为0.1~2.0dtex。另外,对于构成碳纤维前体的纤维束的单纤维的根数也没有特别限定,从性能以及制造成本的平衡的方面出发,优选为1,000~96,000根。
[0037]
水性液可以在制丝工序的任一阶段附着于碳纤维前体的原料纤维,但优选在拉伸工序前一次性附着。另外,只要为拉伸工序前的阶段,即可在任一阶段进行附着。例如可以在刚纺丝后进行附着。进而还可以在拉伸工序后的任一阶段再次进行附着。例如,可以在拉伸工序后立即进行再次附着,可以在卷绕阶段进行再次附着,也可以在即将进行耐火处理工序之前进行再次附着。制丝工序中,进行附着的次数没有特别限定。另外,在水性液附着于碳纤维前体后,可以根据需要进行干燥处理。在附着处理工序后使来自水性液的水分蒸发,使碳纤维用处理剂附着于碳纤维。
[0038]
使第1实施方式的碳纤维前体用处理剂附着于碳纤维前体的比例没有特别限制,优选按照碳纤维前体用处理剂(不包含溶剂)相对于碳纤维前体为0.1~2质量%的方式进行附着,更优选按照为0.3~1.2质量%的方式进行附着。通过为该构成,可进一步提高本发明的效果。作为第1实施方式的水性液的附着方法可以应用公知的方法,其中可以举出例如喷雾给油法、浸渍给油法、辊给油法、使用计量泵的导引给油法等。
[0039]
对本实施方式的水性液和碳纤维前体的作用和效果进行说明。
[0040]
(1)本实施方式中,在制备包含氨基改性硅酮和水的水性液时,使用具有特定分子量分布的非离子表面活性剂。因此能够提高水性液的经时稳定性。特别可提高夏季高温时的经时稳定性。另外可提高由被赋予该水性液而得到的碳纤维前体合成出的碳纤维的强度。
[0041]
需要说明的是,上述实施方式可以如下进行变更来实施。上述实施方式和下述变更例可以在技术上不矛盾的范围内相互组合来实施。
[0042]
·
本实施方式的水性液中,可以在不妨碍本发明效果的范围内进一步混配用于保持水性液的品质的稳定化剂、电荷控制剂、增粘剂、抗氧化剂、防腐剂、抗菌剂、ph调节剂、紫外线吸收剂、其他表面活性剂、其他硅酮类等在碳纤维前体用处理剂中通常使用的成分。实施例
[0043]
以下为了更具体地说明本发明的构成和效果,举出实施例等,但本发明并不限于这些实施例。需要说明的是,以下的实施例和比较例的说明中,份是指质量份,并且%是指
质量%。
[0044]
试验分组1(碳纤维前体用处理剂的水性液的制造)
·
非离子表面活性剂(n
‑
1)的制造方法向高压釜内加入作为构成上述化3的r1的原料醇的癸醇20份、十一烷醇50份、十二烷醇30份,进一步加入氢氧化钾0.05份,将气氛用氮气置换。在150℃缓慢地加入环氧乙烷76份,进行醚化反应。对氢氧化钾进行吸附处理后,进行过滤,由此合成出非离子表面活性剂(n
‑
1)。
[0045]
关于构成化3所表示的非离子表面活性剂(n
‑
1)的各结构(r1、ao、r2),分别示于表1的“化3中的r1的烷基的碳原子数”栏、“r1的原料混合比”栏、“化3中的ao”栏、“化3中的r2的种类”栏。
[0046]
·
非离子表面活性剂(n
‑
2)~(n
‑
11)的制造方法基于下述表1的“化3中的r1的烷基的碳原子数”栏、“r1的原料混合比”栏、“化3中的ao”栏、以及“化3中的r2的种类”栏,利用与上述同样的配方合成非离子表面活性剂(n
‑
2)~(n
‑
11)。
[0047]
关于构成化3所表示的非离子表面活性剂(n
‑
2)~(n
‑
11)的各结构(r1、ao、r2),分别示于表1的“化3中的r1的烷基的碳原子数”栏、“r1的原料混合比”栏、“化3中的ao”栏、“化3中的r2的种类”栏。
[0048]
(实施例1)将(a)氨基改性硅酮(si
‑
1)30份、(b)非离子表面活性剂(n
‑
4)5份以及离子交换水65份充分搅拌后,使用均化器进行乳化,由此制备出实施例1的碳纤维前体用处理剂的固体成分浓度35%水性液。
[0049]
(实施例2~16、比较例1~4)基于表3的各“质量份”栏中记载的(a)氨基改性硅酮、(b)非离子表面活性剂、以及水的各混配量,利用与实施例1相同的配方制备实施例2~16和比较例1~4的各碳纤维前体用处理剂的水性液。
[0050]
将各实施例和比较例中使用的硅酮(si
‑
1)~(si
‑
5)、(rsi
‑
6)、(rsi
‑
7)的结构和运动粘度示于表2的“硅酮结构”栏和“运动粘度”栏中。
[0051]
将各实施例和比较例中使用的(a)氨基改性硅酮和(b)非离子表面活性剂的种类示于表3的各成分的“种类”栏中。另外,将(a)氨基改性硅酮、(b)非离子表面活性剂以及水的水性液中的各混配量示于表3的各成分的“质量份”栏中。另外,将各水性液中的(a)氨基改性硅酮和(b)非离子表面活性剂的合计质量、以及(a)硅酮的含量相对于(b)非离子表面活性剂的含量的质量比分别示于表3的“a成分和b成分的合计质量”栏、以及“a成分/b成分的质量比”栏中。
[0052]
·
分子量分布的测定方法非离子表面活性剂的分子量分布通过以下所示的方法求出。
[0053]
首先,将混配在各实施例和比较例的水性液中的非离子表面活性剂的混合物0.02g采集到西林瓶中,加入四氢呋喃(thf)30ml进行稀释,得到试样溶液。将该试样溶液1ml使用安装有gpc用过滤器的注射器在gpc用试样瓶中除去异物,由此得到试样溶液。使用安装有作为参比柱的tskgel superh
‑
rc、作为测定用柱的tskguardcolumn superh
‑
l、
tskgel superh4000、tskgel superh3000、tskgel superh2000的东曹公司制造的hlc
‑
8320gpc进行gpc测定。关于数均分子量(=mn)和质量平均分子量(=mw),使用作为标准试样的tskgel标准聚苯乙烯制作校正曲线,求出混配在各实施例和比较例的水性液中的非离子表面活性剂的混合物的mn和mw。使用该数值计算出分子量分布(=mw/mn)。将分子量分布的结果示于表3的“分子量分布”栏中。
[0054]
[表1]表1的分组栏中记载的n
‑
1~n
‑
11的各非离子表面活性剂的详细内容以下所述。
[0055]
n
‑
1:聚氧乙烯(n=3:表示环氧乙烷的加成摩尔数。以下相同)癸基醚、聚氧乙烯(n=3)十一烷基醚以及聚氧乙烯(n=3)十二烷基醚的混合物n
‑
2:聚氧乙烯(n=9)癸基醚、聚氧乙烯(n=9)十一烷基醚以及聚氧乙烯(n=9)十二烷基醚的混合物n
‑
3:聚氧乙烯(n=12)癸基醚、聚氧乙烯(n=12)十一烷基醚以及聚氧乙烯(n=12)十二烷基醚的混合物n
‑
4:聚氧乙烯(n=5)十二烷基醚以及聚氧乙烯(n=5)十三烷基醚的混合物n
‑
5:聚氧乙烯(n=10)十二烷基醚以及聚氧乙烯(n=10)十三烷基醚的混合物n
‑
6:聚氧乙烯(n=25)十二烷基醚以及聚氧乙烯(n=25)十四烷基醚的混合物n
‑
7:聚氧乙烯(n=15)聚氧丙烯(m=10:表示环氧丙烷的加成摩尔数。以下相同)十四烷基醚以及聚氧乙烯(n=15)聚氧丙烯(m=10)十五烷基醚的混合物n
‑
8:聚氧乙烯(n=40)聚氧丙烯(m=10)十四烷基醚以及聚氧乙烯(n=40)聚氧丙烯(m=10)十五烷基醚的混合物n
‑
9:聚氧乙烯(n=5)十六烷基乙醚、以及聚氧乙烯(n=5)十八烷基乙醚的混合物n
‑
10:聚氧乙烯(n=7)辛基醚
n
‑
11:聚氧乙烯(n=5)聚氧丙烯(m=5)丁基醚
[0056]
[表2]
[0057]
[表3]
试验分组2(碳纤维前体和碳纤维的制造)使用试验分组1中制备的碳纤维前体用处理剂的水性液,制造碳纤维前体和碳纤维。
[0058]
将由丙烯腈95%、丙烯酸甲酯3.5%、甲基丙烯酸1.5%形成的特性粘度1.80的共聚物溶解在二甲基乙酰胺(dmac)中,制作聚合物浓度为21.0%、60℃时的粘度为500泊的纺丝原液。将纺丝原液利用孔径(内径)0.075mm、孔数12,000的喷丝头以牵伸比0.8吐出到保持在纺浴温度35℃的dmac的70%水溶液的凝固浴中。
[0059]
将凝固丝在水洗槽中脱溶剂并同时进行5倍拉伸,制作水溶胀状态的丙烯酸纤维线料。将其利用将试验分组1中制备的碳纤维前体用处理剂的水性液进一步稀释得到的4%离子交换水溶液通过浸渍法按照碳纤维前体用处理剂的固体成分附着量为1%(不包含溶剂)的方式进行给油。其后将该丙烯酸纤维线料利用130℃的加热辊进行干燥致密化处理,进一步在170℃的加热辊间实施1.7倍的拉伸,之后卷绕在丝管上,由此得到碳纤维前体。将丝由该碳纤维前体解舒,利用具有230~270℃的温度梯度的耐火炉在空气气氛下进行1小
时耐火处理后,连续地在氮气气氛下利用具有300~1,300℃的温度梯度的碳化炉进行烧制,转换成碳纤维,之后卷绕在丝管上。
[0060]
按如下所示评价碳纤维前体用处理剂的水性液的经时稳定性、碳纤维的强度。
[0061]
试验分组3(评价)
·
乳化稳定性的评价将碳纤维前体用处理剂的水性液100ml保存在透明的密闭容器中,在40℃静置3天后,将容器振荡10次,再次在40℃静置3天。目视观察静置后的外观。另外,将保存后的处理剂进一步用离子交换水进行稀释以使得固体成分为5%,目视观察稀释后的外观。按下述基准进行评价,将结果示于表3的“经时稳定性”栏中。
[0062]
·
乳化稳定性的评价基准
◎
(优):几乎未观察到分离、沉淀,外观均匀。另外,在稀释后保持了良好的乳化性。
[0063]
○
(良):稍微观察到乳油化或分离,但乳化性良好,为实用上没有问题的水平。另外,在稀释后保持了良好的乳化性。
[0064]
×
(不良):观察到明显的乳油化或分离。或者在稀释后产生了沉淀或分离。
[0065]
·
碳纤维强度的评价依据jis r7606(对应国际标准iso 11566:1996)对于上述得到的碳纤维的强度进行测定,按下述基准进行评价。将结果示于表3的“强度”栏中。
[0066]
·
碳纤维强度的评价基准
○
(优):3.3gpa以上。
[0067]
×
(不良):小于3.3gpa。
[0068]
-:由于稳定性不良,因此未进行评价。
[0069]
由上述表3的结果也可知,本发明显示出了能够提高碳纤维前体用处理剂的水性液的经时稳定性、并且能够提高碳纤维的强度的效果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1