一种聚酰亚胺纤维膜及其制备方法和应用与流程
1.本发明涉及功能性纤维膜材料技术领域,尤其涉及一种聚酰亚胺纤维膜及其制备方法和应用。
背景技术:
2.近年来,高性能聚合物超细纤维膜材料在光电器件领域中的应用得到了越来越广泛的重视。尤其是兼具耐高温、高反射率、高疏水以及耐紫外辐照特性的聚合物超细纤维膜更是在先进光电领域中受到了广泛关注。例如,具有上述特性的聚合物超细纤维膜在发光二级管(led)反射杯领域具有广泛的应用前景。
3.聚酰亚胺(pi)是一类具有优良耐热稳定性的有机高分子材料,因此在高性能超细纤维膜领域具有潜在的应用。但传统的pi材料由于其分子结构的高极性与高共轭特性,因此通常是难溶难熔的,只能通过其可溶性前驱体-聚酰胺酸(paa)进行超细纤维膜的制造,然后将制备的paa纤维膜在高达300℃以上的高温环境中进行化学亚胺化脱水,进而制得最终的pi纤维膜。该过程存在诸多缺陷,包括:1)paa高温脱水转化为pi的过程中,纤维膜的颜色往往会急剧加深,最终纤维膜的反射率较低;2)paa高温化学脱水转化为pi的过程中,纤维膜内部不可避免地会发生粘连,最终造成纤维膜的反射率下降。此外,常规pi材料由于分子结构中存在较高含量的极性酰亚胺基团,因此其表面通常表现出较强的亲水特性,同时常规pi材料的耐紫外(uv)辐照能力较差。综上所述,常规型pi超细纤维膜虽然具有良好的耐热稳定性,但其疏水性、反射率以及耐uv方面均难以满足led等实际应用的需求。
技术实现要素:
4.本发明的目的在于提供一种聚酰亚胺纤维膜及其制备方法和应用,所制备的聚酰亚胺纤维膜具有高疏水、高耐热、高反射率和耐uv辐照性能。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种聚酰亚胺纤维膜的制备方法,包括以下步骤:
7.将聚酰亚胺树脂、抗紫外线助剂和有机溶剂混合,进行复合,得到纺丝液;
8.将所述纺丝液进行静电纺丝,得到聚酰亚胺纤维膜;
9.所述聚酰亚胺树脂具有式i所示结构:
[0010][0011]
式i中,n=1~500且n为整数;
[0012]-x-为
[0013]
优选的,所述聚酰亚胺树脂包括:
[0014]
n=110、n=79、n=343、n=111n=146。
[0015]
优选的,所述抗紫外线助剂包括紫外线吸收剂、激发态淬灭剂、氢过氧化物分解剂或自由基捕获剂。
[0016]
优选的,所述紫外线吸收剂包括邻羟基二苯甲酮、邻羟基苯并三唑或邻羟基苯并三嗪;所述激发态淬灭剂包括过渡金属有机配合物;所述自由基捕获剂包括受阻胺。
[0017]
优选的,所述静电纺丝的条件包括:喷丝头内径为0.21~0.50mm;电压为12-20kv:推注速度为0.1ml/h;喷丝板与接收装置之间的距离为10~20cm;相对湿度为30
±
10%。
[0018]
优选的,所述抗紫外线助剂在聚酰亚胺纤维膜中的含量为0.1~10wt%。
[0019]
优选的,所述聚酰亚胺树脂的制备方法包括以下步骤:
[0020]
将二胺单体、六氟二酐和非质子极性溶剂混合,进行聚合反应,得到聚酰胺酸溶液;所述二胺单体为3,5-二胺基三氟甲苯、2-三氟甲基-4-胺基-3'-三氟甲基二苯醚、1,4-双[(2-三氟甲基-4-胺基)苯氧基]苯、4,4-双[(2-三氟甲基-4-胺基)苯氧基]联苯或4'-特丁基环己基-3,5-二胺基苯甲酸酯;
[0021]
将所述聚酰胺酸溶液、乙酸酐和吡啶混合,进行酰亚胺化反应,得到聚酰亚胺树脂。
[0022]
优选的,所述二胺单体与六氟二酐的摩尔比为(0.95~1.02):(1.02~0.95);所述聚合反应的温度为0~30℃,时间为10~48h。
[0023]
本发明提供了上述技术方案所述制备方法制备得到的聚酰亚胺纤维膜,包括聚酰亚胺膜基体和分散于所述聚酰亚胺纤维膜基体中的抗紫外线助剂。
[0024]
本发明提供了上述技术方案所述聚酰亚胺纤维膜在光电器件、航空航天、可穿戴显示或汽车领域中的应用。
[0025]
本发明提供了一种聚酰亚胺纤维膜的制备方法,本发明将特定结构的聚酰亚胺树脂作为基体,与抗紫外线助剂复合进行静电纺丝制备聚酰亚胺纤维膜;所用聚酰亚胺树脂含有具有良好疏水特征的含氟基团(如三氟甲基、六氟异丙基);或者引入叔丁基、环己基或酯键,改善现有pi纤维膜亲水性强的技术缺陷。同时,含氟基团具有良好的耐热稳定性,可保证pi纤维膜良好的耐热性能。
[0026]
此外,含氟基团、叔丁基或环己基引入pi纤维膜结构中,可有效降低pi分子链的极性,增加其分子链的摩尔体积,从而有利于改善pi纤维膜的白度,进而提高其光反射率;同时本发明将抗紫外稳定助剂引入pi纤维膜中,从而赋予其良好的抗uv辐照能力。因此,本发明制备的pi纤维膜兼具优良的疏水性、耐热稳定性、高反射率以及抗uv辐照特征,可作为组件应用于led照明、航空航天、光电器件、微电子(如发光二级管(led))、可穿戴显示以及汽车等高技术领域,克服了已有pi纤维膜亲水性强、反射率低、耐uv稳定性差等缺陷。
[0027]
本发明所用聚酰亚胺树脂中,含有高电负性以及庞大自由体积的含氟基团(如三氟甲基或六氟异丙基);或者叔丁基、环己基等引入pi纤维膜结构中,一方面赋予pi树脂在有机溶剂中良好的溶解性,进而开发有机可溶性pi,使pi树脂溶解于极性非质子性溶剂中配制成预亚胺化的pi溶液,进而通过静电纺丝可直接制得pi纤维膜,从而避免了传统采用paa溶液进行电纺,然后在高温下进行化学脱水转化为pi纤维膜的技术缺陷。
[0028]
本发明的制备方法简单且高效,收率高。
附图说明
[0029]
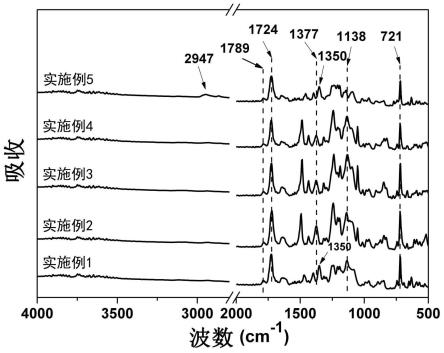
图1为实施例1~5制备所得pi树脂的红外谱图;
[0030]
图2为实施例1~5制备所得pi树脂的核磁氢谱;
[0031]
图3为实施例1~5制备所得pi纤维膜的x射线衍射图;
[0032]
图4为实施例1~5制备所得pi纤维膜的扫描电镜图和粒径分布图;
[0033]
图5为实施例1~5制备所得pi纤维膜的紫外-可见光谱;
[0034]
图6为实施例1~5制备所得pi纤维膜的热失重谱图;
[0035]
图7为实施例1~5制备所得pi纤维膜的差示扫描量热谱图。
具体实施方式
[0036]
本发明提供了一种聚酰亚胺纤维膜的制备方法,包括以下步骤:
[0037]
将聚酰亚胺树脂、抗紫外线助剂和有机溶剂混合,进行复合,得到纺丝液;
[0038]
将所述纺丝液进行静电纺丝,得到聚酰亚胺纤维膜;
[0039]
所述聚酰亚胺树脂具有式i所示结构:
[0040][0041]
式i中,n=1~500且n为整数;
[0042]-x-为为
[0043]
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
[0044]
本发明将聚酰亚胺树脂、抗紫外线助剂和有机溶剂混合,进行复合,得到纺丝液。
[0045]
在本发明中,所述聚酰亚胺树脂具有式i所示结构:
[0046][0047]
式i中,n=1~500且n为整数;
[0048]-x-为为
[0049]
在本发明中,所述n优选为10~400;所述聚酰亚胺树脂优选包括:
[0050]
n=110、n=79、n=343、n=111n=146。
[0051]
在本发明中,所述聚酰亚胺树脂的制备方法优选包括以下步骤:
[0052]
将二胺单体、六氟二酐和非质子极性溶剂混合,进行聚合反应,得到聚酰胺酸溶液;所述二胺单体为3,5-二胺基三氟甲苯、2-三氟甲基-4-胺基-3'-三氟甲基二苯醚、1,4-双[(2-三氟甲基-4-胺基)苯氧基]苯、4,4-双[(2-三氟甲基-4-胺基)苯氧基]联苯或4'-特丁基环己基-3,5-二胺基苯甲酸酯;
[0053]
将所述聚酰胺酸溶液、乙酸酐和吡啶混合,进行酰亚胺化反应,得到聚酰亚胺树脂。
[0054]
在本发明中,所述二胺单体优选为3,5-二胺基三氟甲苯(tfmda)、2-三氟甲基-4-胺基-3'-三氟甲基二苯醚(tfoda)、1,4-双[(2-三氟甲基-4-胺基)苯氧基]苯(6fapb)、4,4-双[(2-三氟甲基-4-胺基)苯氧基]联苯(6fbab)或4'-特丁基环己基-3,5-二胺基苯甲酸酯(dabc)。
[0055]
在本发明中,所述非质子极性溶剂优选为n-甲基吡咯烷酮(nmp)、间甲酚、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、二甲基亚砜(dmso)和γ-丁内酯中的至少一种,更优选为n,n-二甲基乙酰胺;当所述非质子极性溶剂为上述中的几种时,本发明对不同种类非质子极性有机溶剂的配比没有特殊的限定,任意配比均可;所述非质子极性溶剂的用量优选为使二胺单体、六氟二酐和非质子极性溶剂混合所得反应体系固含量为10~30wt%,更优选为15~25wt%,进一步优选为20wt%;
[0056]
在本发明中,所述二胺单体与六氟二酐(6fda)的摩尔比优选为(0.95~1.02):(1.02~0.95),更优选为(0.98~1.01):1,进一步优选为1:1。
[0057]
在本发明中,所述二胺单体、六氟二酐和非质子极性溶剂混合的过程优选为将二胺单体溶于非质子极性溶剂,在搅拌条件下形成均相溶液,加入六氟二酐,洗涤残留六氟二酐。
[0058]
在本发明中,所述聚合反应优选在氮气条件下进行;所述聚合反应的温度优选为0~30℃,更优选为10~25℃;时间优选为10~48h,更优选为18~24h。
[0059]
完成所述聚合反应后,本发明优选不进行处理,得到聚酰胺酸溶液。
[0060]
在本发明中,所述六氟二酐、乙酸酐和吡啶的摩尔比优选为1:(3~20):(2~16),更优选为1:(5~10):(4~8)。
[0061]
本发明对所述聚酰胺酸溶液、乙酸酐和吡啶混合的过程没有特殊的限定,按照本领域熟知的过程进行即可;所述酰亚胺化反应的温度优选为0~25℃,更优选为15~25℃,时间优选为10~48h,更优选为12~24h。本发明利用乙酸酐作为脱水剂,吡啶作为催化剂。
[0062]
完成所述酰亚胺化反应后,本发明优选将所得产物沉淀到过量无水乙醇中,得到聚酰亚胺树脂。本发明对所述沉淀的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0063]
在本发明中,所述抗紫外线助剂优选包括紫外线吸收剂、激发态淬灭剂、氢过氧化物分解剂或自由基捕获剂;所述紫外线吸收剂优选包括邻羟基二苯甲酮、邻羟基苯并三唑或邻羟基苯并三嗪;所述激发态淬灭剂优选包括过渡金属有机配合物,所述过渡金属有机配合物优选为镍有机络合物;所述镍有机络合物优选为市售uv-1084化合物;所述氢过氧化物分解剂优选包括氨基甲酸酯或亚膦酸酯;所述自由基捕获剂优选包括受阻胺。
[0064]
在本发明中,所述有机溶剂优选为n-甲基吡咯烷酮(nmp)、n,n-二甲基乙酰胺
(dmac)、二甲基亚砜(dmso)和n,n-二甲基甲酰胺(dmf)中的一种或几种;当所述有机溶剂为上述中的几种时,本发明对不同种类有机溶剂的配比没有特殊的限定,任意配比均可。
[0065]
在本发明中,所述聚酰亚胺树脂、抗紫外线助剂和有机溶剂混合的过程优选为将聚酰亚胺树脂分散于部分有机溶剂,得到聚酰亚胺分散液;将抗紫外线助剂分散于剩余有机溶剂,得到助剂分散液;将所述聚酰亚胺分散液和助剂分散液混合。在本发明中,所述聚酰亚胺分散液的固含量优选为15~40wt%,更优选为20wt%;所述助剂分散液的固含量优选为1~10wt%,更优选为2wt%;所述聚酰亚胺分散液与助剂分散液的重量比优选使得抗紫外线助剂在聚酰亚胺纤维膜固体中的含量为0.1~10wt%,更优选为0.5wt%。
[0066]
本发明对所述复合的过程没有特殊的限定,在搅拌条件下将物料混合均匀即可;本发明对所述搅拌的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0067]
得到纺丝液后,本发明将所述纺丝液进行静电纺丝,得到聚酰亚胺纤维膜。
[0068]
在本发明中,所述静电纺丝的条件优选包括:喷丝头内径为0.21~0.50mm;电压为12~20kv,更优选为15kv:推注速度为0.1ml/h;喷丝板与接收装置之间的距离为10~20cm,更优选为15cm;相对湿度为30
±
10%。
[0069]
完成所述静电纺丝后,本发明优选还包括将所得膜进行热处理,得到聚酰亚胺纤维膜;所述热处理的温度优选为180~200℃,时间优选为0.5~5h,更优选为1~3h;本发明通过热处理去除膜的残留溶剂,得到聚酰亚胺纤维膜。
[0070]
本发明提供了上述技术方案所述制备方法制备得到的聚酰亚胺纤维膜,包括聚酰亚胺膜基体和分散于所述聚酰亚胺纤维膜基体中的抗紫外线助剂。
[0071]
在本发明中,所述聚酰亚胺纤维膜中单根纤维的平均直径优选为200~2000nm。
[0072]
本发明提供了上述技术方案所述聚酰亚胺纤维膜在光电器件、航空航天、可穿戴显示或汽车领域中的应用。在本发明中,所述光电器件优选包括发光二极管。本发明对所述应用的方法没有特殊的限定,按照本领域熟知的方法应用即可。
[0073]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0074]
实施例1
[0075]
由6fda与tfmda制备pi纤维膜:
[0076]
pi树脂合成:在配有机械搅拌、温度计和氮气入口的250ml三口瓶中,将tfmda(3.5228g,0.02mol)溶于新蒸馏的dmac(40.0g)中,得到澄清的二胺溶液;在二胺溶液中加入6fda(8.8848g,0.02mol),并加入另外体积的dmac(9.6g)洗涤残留的二酐,同时将反应体系的固含量调节至20wt%;在25℃下,在氮气中搅拌24h后,得到聚酰胺酸(paa)溶液,向所述聚酰胺酸溶液中加入乙酸酐(10.209g,0.1mol)和吡啶(6.328g,0.08mol)的混合物,将反应混合物在25℃下搅拌24h,将所得粘稠溶液缓慢倒入过量的乙醇中,得到白色纤维状聚酰亚胺树脂,即pi树脂,结构如下所示:
[0077]
[0078]
该pi树脂的数均分子量(mn)为64505g/mol,重均分子量(mw)为116251g/mol;n=110。
[0079]
pi纤维膜的制备:将所述聚酰亚胺树脂溶解于n,n-二甲基乙酰胺(dmac)中,配制成固含量为20wt%的溶液a;将受阻胺类uv光稳定剂(2020,德国basf产品)溶解于dmac中,配制成固含量为2wt%的溶液b;取溶液a4g,溶液b 0.2g进行充分混合,得到纺丝溶液,采用该纺丝溶液进行静电纺丝,纺丝参数如下:喷丝头内径0.50mm;施加电压15kv;推注速度0.1ml/h;喷丝板与接收装置之间的距离为15cm;相对湿度为30
±
10%,将纺丝所得膜在200℃下干燥3h,得到聚酰亚胺纤维膜中,其中,抗紫外线助剂的质量百分含量为0.5wt%。
[0080]
实施例2
[0081]
由6fda与tfoda制备pi纤维膜:
[0082]
pi树脂合成步骤同实施例1,区别在于:将tfmda(3.5228g,0.02mol)替换为tfoda(5.3646g,0.02mol),所得pi树脂的结构如下所示:
[0083][0084]
该pi数值的数均分子量(mn)为64505g/mol,重均分子量(mw)为116251g/mol;n=79。
[0085]
pi纤维膜的制备步骤同实施例1,所制备的pi纤维膜中,抗紫外线助剂的质量百分含量为0.5wt%。
[0086]
实施例3
[0087]
由6fda与6fapb制备pi纤维膜:
[0088]
pi树脂合成步骤同实施例1,区别在于:将tfmda(3.5228g,0.02mol)替换为6fapb(8.5666g,0.02mol),所得pi树脂的结构如下所示:
[0089][0090]
该pi树脂的数均分子量(mn)为287038g/mol,重均分子量(mw)为474745g/mol;n=343。
[0091]
pi纤维膜的制备步骤同实施例1,所制备的pi纤维膜中,抗紫外线助剂的质量百分含量为0.5wt%。
[0092]
实施例4
[0093]
由6fda与6fbab制备pi纤维膜:
[0094]
pi树脂合成步骤同实施例1,区别在于:将tfmda(3.5228g,0.02mol)替换为6fbab(10.0884g,0.02mol),所得pi树脂的结构如下所示:
[0095][0096]
该pi数值的数均分子量(mn)为101235g/mol,重均分子量(mw)为204765g/mol;n=111。
[0097]
pi纤维膜的制备步骤如实施例1,所制备的pi纤维膜中,抗紫外线助剂的质量百分含量为0.5wt%。
[0098]
实施例5
[0099]
由6fda与dabc制备pi纤维膜:
[0100]
pi树脂合成步骤同实施例1,区别在于:将tfmda(3.5228g,0.02mol)替换为dabc(5.8080g,0.02mol),所得pi树脂的结构如下所示:
[0101][0102]
该pi树脂的数均分子量(mn)为102064g/mol,重均分子量(mw)为176971g/mol;n=146。
[0103]
pi超细纤维膜的制备步骤同实施例1,所制备的pi纤维膜中,抗紫外线助剂的质量百分含量为0.5wt%。
[0104]
对比例1
[0105]
由pmda与4,4
′‑
二胺基二苯醚(oda)制备pi纤维膜:
[0106]
在配有机械搅拌、温度计和氮气入口的250ml三口瓶中,将oda(2.0024g,0.01mol)溶于新蒸馏的dmac(30.6g)中,得到澄清的二胺溶液;在二胺溶液中加入pmda(2.1812g,0.01mol),并加入另外体积的dmac(7.0g)洗涤残留的二酐,同时将反应体系的固体含量调节至10wt%,室温下,在氮气中搅拌5h后,得到聚酰胺酸溶液;
[0107]
将所述聚酰胺酸溶液稀释于n,n-二甲基乙酰胺(dmac)中,配制成固含量为8wt%的溶液进行静电纺丝,纺丝参数如下:喷丝头内径0.50mm;施加电压15kv:推注速度0.1ml/h;喷丝板与接收装置之间的距离为15cm;相对湿度为30
±
10%,得到paa纤维膜;
[0108]
将所述paa纤维膜在300℃下处理1h,得到pi纤维膜,该pi纤维膜中pi树脂的结构如下所示:
[0109][0110]
对比例2
[0111]
由6fda与oda制备pi纤维膜:
[0112]
在配有机械搅拌、温度计和氮气入口的250ml三口瓶中,将oda(2.0024g,0.01mol)
溶于新蒸馏的dmac(20.8g)中,得到澄清的二胺溶液;在二胺溶液中加入6fda(4.4424g,0.01mol),并加入另外体积的dmac(5.0g)洗涤残留的二酐,同时将反应体系的固体含量调节至20wt%,室温下,在氮气中搅拌5h后,得到聚酰胺酸溶液;
[0113]
将所述聚酰胺酸溶液稀释于n,n-二甲基乙酰胺(dmac)中,配制成固含量为8wt%的溶液进行静电纺丝,纺丝参数如下:喷丝头内径0.50mm;施加电压15kv:推注速度0.1ml/h;喷丝板与接收装置之间的距离为15cm;相对湿度为30
±
10%,得到paa纤维膜;
[0114]
将所述paa纤维膜在300℃下处理1h,得到pi纤维膜,该pi纤维膜中pi树脂的结构如下所示:
[0115][0116]
性能测试
[0117]
1)对实施例1~5和对比例1~2制备的纤维膜进行性能测试,所采用的方法分别为:
[0118]
pi树脂的分子量:
[0119]
凝胶渗透色谱(gpc):将pi树脂通过日本岛津公司的lc-20ad高效液相色谱测试,n-甲基吡咯烷酮(nmp)为流动相,所得分子量均为数均分子量。
[0120]
pi树脂的化学结构:
[0121]
红外光谱(ft-ir):将pi树脂通过德国布鲁克公司的brukertensor-27红外光谱仪测试。
[0122]
核磁氢谱(1h-nmr)采用日本理化的av 400核磁共振谱仪测试,频率为300mhz,氘代二甲基亚砜(dmso-d6)作为核磁试剂。
[0123]
广角x射线衍射采用日本理光的d/max-2500x射线衍射仪测试。
[0124]
pi纤维膜的微观形貌评价方法:
[0125]
扫描电镜(sem):将制备的pi纤维膜在日本jeol公司jsm-it300系列扫描电镜上测试,加速电压:5-20kv。
[0126]
pi纤维膜的水接触角评价方法:
[0127]
采用德国kruess公司的dsa30接触角测试仪测试,水滴量为2μl,测试温度为25
±
0.5℃。
[0128]
pi纤维膜的热分解温度评价方法:
[0129]
热重分析(tga):将制备的pi纤维膜在美国perkinelmer公司sta8000热重分析仪上测试,升温速度:20℃/min,氮气气氛。
[0130]
差示扫描量热分析(dsc):将制备的pi纤维膜在美国ta公司q-100差示扫描量热仪上测试,升温速度:10℃/min,氮气气氛。
[0131]
pi纤维膜的反射率评价方法:
[0132]
紫外-可见光反射光谱(uv-vis):将制备的pi纤维膜在日本hitachi公司u-3900型紫外分光光度计上测试,波长范围200-800nm。r457定义为457nm处样品的反射率。
[0133]
pi纤维膜的耐uv辐照性能评价方法:
[0134]
将pi纤维膜置于氙灯(北京中教金源科技有限公司,cel-hxuv300)下进行辐照,辐照能量为600w/m2,辐照时间为24h;测试辐照前后纤维膜的反射率变化情况。
[0135]
以上性能测试结果见图1~7和表1。
[0136]
图1为实施例1~5制备所得pi树脂的红外谱图;从图1中可以准确指认出酰亚胺环在1789cm-1
、1724cm-1
以及1377cm-1
处的特征吸收峰。
[0137]
图2为实施例1~5制备所得pi树脂的核磁氢谱;由图2可以看出,芳香环h质子的吸收出现在谱图的低场位置(7.5~8.5ppm),其中,6fda单元上的hb质子的吸收出现在了谱图的最低场。实施例5制备的pi纤维膜中,芳香环h质子的吸收出现在谱图的低场位置(7.5~8.5ppm),而脂肪链上的h质子的吸收出现在谱图的高场位置(0.5~2.5ppm)。其中,6fda单元上的hb质子的吸收出现在了谱图的最低场,而dabc单元上的甲基h质子的吸收出现在谱图的最高场。
[0138]
图3为实施例1~5制备所得pi纤维膜的x射线衍射图;由图3可以看出,制备的pi纤维膜在2θ=16
°
左右出现宽大的衍射峰,表明该pi纤维膜的聚集态结构为无定型结构,6fda二酐单体中灵活而庞大的基团有效地阻止了pi中分子链的有序堆积。
[0139]
图4为实施例1~5制备所得pi纤维膜的扫描电镜图和粒径分布图,每个实施例的左侧附图为sem图,右侧为对应的粒径分布图;从图4中可以看出,实施例1制备的pi纤维膜中每根纤维的平均直径为360nm;实施例2制备的pi纤维膜中每根纤维的平均直径为665nm,实施例3制备的pi纤维膜中每根纤维的平均直径为1937nm;实施例4制备的pi纤维膜中每根纤维的平均直径为1037nm;实施例5制备的pi纤维膜中每根纤维的平均直径为1363nm。
[0140]
图5为实施例1~5制备所得pi纤维膜的紫外-可见光谱;由图5可以看出,制备的pi纤维膜在437~760nm波长处的反射率高于80.0%。
[0141]
图6为实施例1~5制备所得pi纤维膜的热失重谱图;由图6可以看出,制备的pi纤维膜在温度达到450℃之前表现出了良好的耐热稳定性。
[0142]
图7为实施例1~5制备所得pi纤维膜的差示扫描量热谱图。由图7可以看出,制备的pi纤维膜在加热过程中表现出明显的玻璃化转变行为,玻璃化转变温度(tg)为297.9℃;实施例2制备的pi纤维膜的玻璃化转变温度(tg)为287.5℃;实施例3制备的pi纤维膜的玻璃化转变温度(tg)为255.4℃;实施例4制备的pi纤维膜的玻璃化转变温度(tg)为272.3℃,实施例5制备的pi纤维膜玻璃化转变温度(tg)为384.7℃。
[0143]
表1实施例1~5和对比例1~2制备的pi纤维膜的性能数据
[0144][0145]
由表1可知,实施例1~5制备的pi树脂可溶解于dmac中,而对比例1与对比例2只能采用paa进行静电纺丝制备纤维膜。paa纤维膜转化为pi纤维膜时,纤维膜发生显著黄变和粘连,造成反射率明显降低。由实施例1~5制备的pi纤维膜的5%失重温度以及玻璃化转变温度数据可以看出,本发明所用pi树脂材料具有优良的耐热稳定性,与对比例1和对比例2制备的材料具有类似的耐热稳定性。
[0146]
由实施例1~5制备的pi纤维膜的水接触角测试结果可以看出,这类材料的水接触角在130~165
°
,显著高于对比例1和对比例2制备的材料的72
°
和93
°
。这主要是由于本发明的pi材料分子结构中引入了疏水性三氟甲基或酯键和环己烷等基团的缘故。
[0147]
由实施例1~5制备的pi纤维膜的反射率测试结果可以看出,这类材料在457nm波长处的反射率在71.9~92.5%之间,显著高于对比例1和对比例2制备的材料的30.4%和46.2%。这主要是由于本发明的pi材料分子结构中引入了高电负性三氟甲基或低共轭环己烷等基团的缘故。
[0148]
由实施例1~5制备的pi纤维膜经uv辐照后的反射率测试结果可以看出,这类材料在uv辐照后,在457nm波长处的反射率降低程度较低,而同样条件下,对比例1和对比例2制备的材料经uv辐照后,反射率发射率大幅度的衰减。这主要是由于本发明的pi分子结构中引入了耐ub辐照的三氟甲基或低共轭环己烷等基团,同时加入了抗紫外线助剂的缘故。
[0149]
综上,本发明提供的pi纤维膜材料具有优良的综合性能,包括高疏水、高耐热、高反射率和耐uv辐照,综合性能优于现有pi纤维膜,具有良好的工业化前景。
[0150]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1