一种数据库的建立方法和遗传疾病的风险预测方法与流程
本发明涉及一种生物
技术领域:
,尤其涉及一种数据库的建立方法和遗传疾病的风险预测方法。
背景技术:
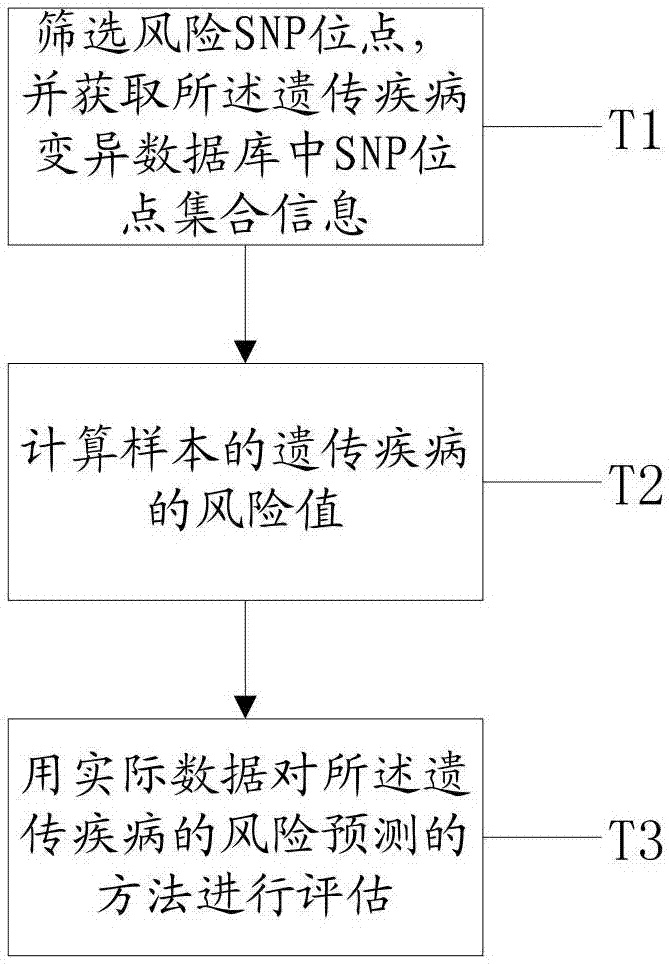
:目前,复杂疾病,如:胃癌、结肠癌等疾病的遗传不遵循孟德尔遗传模式,其发生受多个微效基因及环境因素的影响。复杂疾病在全球范围内广泛流行,严重危害人类的健康,人们迫切希望从根本上找到这些疾病的发病机理,为疾病的诊断、治疗及预防提供基础和保障。过去十年,随着高通量分子检测技术的发展,已经发现了很多疾病相关基因变异位点,尤其是全基因组关联研究(GWAS)技术的发展,加快了人类常见复杂疾病的研究步伐,发现了很多疾病相关的风险位点(SNP),这些信息的发现为预测疾病风险提供了一种可能。通过预测疾病风险,可以提前预警,督促改变(如:生活方式改变),为个性化医疗,个性化健康管理提供了一种策略。从长远角度看,个性化医疗通过更精确的诊断,预测潜在疾病的风险,提供更有效、更有针对性的治疗,预防某种疾病的发生,防患于未然比“治有病”更节约治疗成本。GWAS被广泛应用于复杂疾病的遗传学研究并取得了一系列成果,但是GWAS的位点信息很多都是通过文献获得,所以,构建一个信息比较全面的复杂疾病数据GWAS数据库尤其显得重要。目前,收集整理GWAS信息的数据库最主有Clinvar和GWAS,Clinvar是一个公开的数据库,其中收集了与疾病相关的遗传变异,包含GWAS中部分重要位点信息。GWAS数据库中收集了与疾病相关的遗传变异。尽管这些数据库收集了大量的疾病相关的位点信息,但是这些信息来源多样,包含着大量的噪音,不能直接用于疾病风险的预测,因为:1,疾病名称没有标准化,由于来源于不同的文献,很多是人工收集,所以,即使是同一种表型,其命名方式也多种多样。2,重要信息不全,数据库信息不全,如进行风险预测的时,需要确定风险等位基因型和OR值。所以,我们对Clinvar数据的疾病名称进行标准化,并对Clinvar数据库中的重要信息进行补充,构建了本地复杂疾病数据库。复杂疾病GWAS构建好以后,GWAS还面临着一些挑战,比如:复杂疾病相关基因位点数目众多,每一个位点所起到的作用大小不同。GRS(Geneticriskscore,遗传风险评分)能整合多个SNPs的综合信息来评价基因序列变异和疾病之间的联系。GRS的构建基于多基因模型,假定疾病的遗传效应等于各个位点的效应之和,算法分两种:简单的GRS和加权的GRS。其中加权GRS更接近于真实事实,该算法认为每个风险等位基因对疾病的影响不同,通过给每个风险等位基因赋予一个相应的权重来显示不同SNPs对疾病的影响程度不同。鉴于此,目前,还没有很好的信息比较全面的复杂疾病变异数据库,没有针对遗传风险评估方法。针对现有技术中所存在的问题,提供一种数据库的建立方法和遗传疾病的风险预测方法具有重要意义。技术实现要素:为解决上述问题,本发明提供一种数据库的建立方法和遗传疾病的风险预测方法。为实现上述目的,本发明的一种数据库的建立方法,基于第一数据和第二数据建立第一数据库;根据所述第一数据库中的第一属性对所述第一数据库进行分类,选择分类后的第三数据;将所述第二数据与所述第三数据合并,生成整合数据;根据所述整合数据建立第二数据库;进一步地,将所述第二数据与所述第三数据合并,生成整合数据之后,还包括:补充所述整合数据中的缺失数据;进一步地,基于第一数据和第二数据建立第一数据库之后,还包括:对所述第一数据和第二数据进行正则化;进一步地,所述正则化具体为,用python的正则表达式和文本处理包对数据进行正则化;进一步地,所述数据库为一种遗传疾病变异数据库,所述第一数据为Clinvar数据,所述第二数据为GWAS数据,所述方法包括:基于Clinvar数据和GWAS数据建立第一数据库;对所述Clinvar数据和所述GWAS数据进行正则化;根据所述Clinvar数据库中的Clinicalsignificance属性对所述第一数据库进行分类,选择分类后的GWAS,riskfactor和protective三类数据作为第三数据;对所述第三数据进行正则化;将所述GWAS数据与所述第三数据合并,生成整合数据;补充所述整合数据中的缺失数据;根据所述整合数据建立所述遗传疾病变异数据库;本发明还提供了一种遗传疾病的风险预测方法,所述方法基于遗传疾病变异数据库,所述方法包括:筛选风险SNP位点,并获取所述遗传疾病变异数据库中SNP位点集合信息;计算样本的遗传疾病的风险值;进一步地,所述计算样本的遗传疾病的风险值之后,还包括:用实际数据对计算结果进行评估;进一步地,所述计算样本的遗传疾病的风险值,具体为:所述a为疾病的发病率;所述s为基因名称;所述OR为每个SNP位点的比值比;所述WORi(s,OR)为每个SNP位点加权后的比值比;所述为样本的遗传疾病的风险值。本发明的一种数据库的建立方法和遗传疾病的风险预测方法构建了遗传疾病变异数据库,并基于所述遗传疾病变异数据库结合加权GRS方法和贝叶斯公式构建了遗传疾病的风险预测的方法,使得所述遗传疾病变异数据库的记录的信息和文字标准化的同时,实现对遗传疾病的风险的准确预测。附图说明图1为本发明所述数据库的建立方法的流程示意图;图2为本发明所述遗传疾病的风险预测方法的流程示意图。具体实施方式下面,结合附图,对本发明的结构以及工作原理等作进一步的说明。如图1所示,图1为本发明所述数据库的建立方法的流程示意图,包括:S1基于第一数据和第二数据建立第一数据库;通常地,用mysql基于第一数据和第二数据建立第一数据库。S2对所述第一数据和第二数据进行正则化;S3根据所述第一数据库中的第一属性对所述第一数据库进行分类,选择分类后的第三数据;S4将所述第二数据与所述第三数据合并,生成整合数据;S5补充所述整合数据中的缺失数据;在本发明优选的实施例中,所述正则化具体为用python的正则表达式和文本处理包对数据进行正则化。正则表达式是用于处理字符串的工具,通常地,正则表达式的匹配过程是:拿出表达式和文本中的字符比较,若每一个字符都能匹配,则匹配成功;若存在匹配不成功的字符则匹配失败。Python作为一种面向对象的解释型计算机程序设计语言,提供正则表达式模式,并拥有全部的正则表达式功能。Python通过re模块提供对正则表达式的支持。所述文本处理包(NaturalLanguageToolkit)为在NLP领域中,最常使用的Python库。因此,用所述python的正则表达式和文本处理包能够实现对数据的正则化及标准化。在本发明的实施例一中,所述数据库的建立方法具体为一种遗传疾病变异数据库的建立方法,所述第一数据为Clinvar数据,所述第二数据为GWAS数据,所述Clinvar是一个以美国医学遗传学与基因组学学会(ACMG),临床药物基因组学实施联盟(CPIC)等为依据的公开的数据库,其中收集了与疾病相关的遗传变异,所述Clinvar数据即为所述Clinvar数据库中的原始数据,所述Clinvar数据库中的原始数据可以通过NCBI下载得到。所述GWAS即全基因组关联分析,是指在人类全基因组范围内找出存在的序列变异,即单核苷酸多态性(SNP),从中筛选出与疾病相关的SNPs,所述GWAS数据即为GWAS的原始数据,所述GWAS的原始数据可以从GWAS的官网下载得到。所述遗传疾病变异数据库的建立方法具体为:用mysql基于Clinvar数据和GWAS数据建立第一数据库;解析所述Clinvar数据文本和所述GWAS数据文本,并用python的正则表达式和文本处理包,对所述Clinvar数据和所述GWAS数据进行正则化;所述Clinvar数据作为Clinvar的原始数据,以每个变异作为行记录单元,所以,每行会出现一种变异对应多种表型、疾病的列表,多种临床显著性,即(1个)snp对应(1个或多个)表型对应(1个或多个)临床显著性,在用python的正则表达式和文本处理包进行正则化后,处理成,(1个)snp对应(1个)表型对应(1个)临床显著性。所述GWAS数据的内容中,许多都是自然语言,是非结构化的内容,不易后续的使用,所以,需要对其内容进行规范,因此需要用python的正则表达式和文本处理包对所述GWAS数据进行正则化后,使所述GWAS数据规范化。根据所述Clinvar数据库中的Clinicalsignificance属性对所述第一数据库进行分类,所述分类后共有9类,分别是Mendeliandisorders、Drugresponse、GWAS、riskfactor、protective、non-diseasephenotype、conflict、other、notprovided,选择分类后的GWAS,riskfactor和protective这三类数据作为第三数据,作为遗传疾病变异数据库的数据源之一。将所述GWAS数据与所述第三数据合并,生成整合数据。补充所述整合数据中的缺失数据;根据所述整合数据建立所述遗传疾病变异数据库;在GWAS官网中下载的数据很多都是人工整理的,所以下载数据内容都不全,需要将缺失的数据补充完成。如图2所示,图2为本发明所述遗传疾病的风险预测方法的流程示意图,本发明还提供了一种遗传疾病的风险预测方法,所述方法基于遗传疾病变异数据库,所述方法包括:T1筛选风险SNP位点,并获取所述遗传疾病变异数据库中SNP位点集合信息;T2计算样本的遗传疾病的风险值;T3用实际数据对所述遗传疾病的风险预测的方法进行评估;所述计算样本的遗传疾病的风险值,具体为:所述a为疾病的发病率;所述s为基因名称;所述OR为每个SNP位点的比值比;所述WORi(s,OR)为每个SNP位点加权后的比值比;所述为样本的遗传疾病的风险值。其中,所述WOR的计算方法,具体为:所述WORi(s,OR)表示不同情况下的权重后的OR值;所述s为基因名称;所述OR为每个SNP位点的比值比;所述为基因上的SNP加权后的OR值。其中,所述的计算方法,具体为:所述为基因上的SNP加权后的OR值,所述P(rs,Nrs)为相同的SNP-phenotype在不同文献中同时出现的次数,所述W(s,Ns)为根据相同的Gene-Phenotype在不同文献中同时出现的次数,确定加权程度,如果文献数目小于10,则W(s,Ns)=1,若所述文献数目大于10,则W(s,Ns)=2,所述OR为每个SNP位点的比值比,所述dwOR为根据自有样本库计算出的OR值;所述自有样本库为根据本公式收集的一些已知临床表型的中国人实测样本,计算dwOR值,将这一值作为加权项,并且可以随着收集的样本量不断调整dwOR值。其中W(s,Ns)的具体计算方法解释为,所述Gene-Phenotype为根据文献挖掘所构建的数据库,计算变异所属的基因是否在整个pubmed文献中有多篇报道。若有多篇文献报道,则说明该基因对某一表型起更重要的作用,该基因上发生SNP则影响更大,权重也加大。其中P(rs,Nrs)的具体计算方法解释为,SNP-phenotype为若某一SNP被多篇GWAS文献报道与某一疾病,表型相关,则说明该位点与疾病的关系更可信,所以,相对于文献报道数少的,某一SNP被多篇文献报道,所加的权重更大;其中,所述dwOR的具体计算方法为:所述naa,maa为基因型(genotype)是aa,在疾病组和对照组的样本数量;所述nab,mab为基因型是ab,在疾病组和对照组的样本数量;所述nbb,mbb为基因型是bb,在疾病组和对照组的样本数量;所述dwOR为根据自有样本库计算出的OR值,具体为,根据某一疾病的样本和正常样本计算出的OR值,当OR>1时,则表示该因素是一个危险因素,当OR<1时,则表示该因素是一个保护因素。其中,所述P(a,n,m)的具体计算方法为:所述naa,maa为基因型是aa,在疾病组和对照组的样本数量;所述nab,mab为基因型是ab,在疾病组和对照组的样本数量;所述nbb,mbb为基因型是bb,在疾病组和对照组的样本数量;所述P(a,n,m)为每种基因型在疾病组和对照组的分布频率之比。在本发明的技术方案中,所述用实际数据对计算结果进行评估具体为通过对GSR算法进行测试,计算预测的准确性;在预测结果中,样本输入本身带有标签,阳性(positive)或阴性(negative),GSR算法会对每个样本重新分类为阳性或阴性,如果分类结果为阳性且和输入样本标签一致,称为真阳性(Truepositive,TP),如不一致,称为假阳性(Falsepositive,FP);如分类结果为阴性且与输入时标签一致,称为真阴性(Truenegative,TN),不一致则成为假阴性(Falsenegative,FN)。通常地,衡量算法性能的参数基于四个常用的评估指标:准确率,特异性,灵敏性和马修斯相关系数。其中,所述准确率(Accuracy)为结果中正确预测结果的比例,包括真阳性和真阴性,所述准确率的计算方法为:所述特异性(Specificity)为得出阴性检验结果的阴性样本占所有阴性样本的比例,所述特异性的计算方法为:所述灵敏性(Sensitivity)为得出阳性检验结果的阳性样本占所有阳性样本的比例。灵敏性越高,表示检出阳性样本的能力越强,所述灵敏性的计算方法为:所述马修斯相关系数(Matthewscorrelationcoefficient,MCC)是预测结果和观察结果间的相关性,其取值范围是-1到1,MCC越大,预测性能越好,所述马修斯相关系数的计算方法为:在本发明的实施例一中,需要检测遗传疾病变异数据库中亚洲人群胃癌的风险,则选取所述遗传疾病变异数据库数据库中亚洲人群的胃癌风险SNP位点,构造sample1,如表1所述:将表1中的数据代入公式计算可得在预设的评判标准中,GRS>0.997为高风险,因此可以推断该数据库的样本数据中,表2中的样本是胃癌发生的高风险样本。计算结果如表2所示:snp_idriskORpubmedgenotypesamplescoreGRS2294008T1.6011111184880301/113.20222220.9991242976392A1.6317167184880301/113.26343330.999142920297G1.3263245267018791/112.6526490.9989431045531A1.5203108184880301/113.04062150.99907710216533A1.5263245184880301/113.0526490.9990812976395A1.5263245184880301/113.0526490.999081接下来,用实际数据对所述遗传疾病的风险预测的方法的计算结果进行评估,对30例胃癌样本和203例正常样本进行计算,根据GRS的风险预测结果和实际样本的病理学检测结果进行比较,结果如下表所示:actualpositiveactualnegativepredictedpositive269predictednegative4194可知,TP为26,TN为194,FP为4,FN为9。将结果代入公式中可得准确性为,Accuracy=0.994;代入公式,中可得特异性为,Specificity=0.956;代入公式,中可得灵敏性为,Sensitivity=0.867;代入公式,中可得马修斯相关系数为,MCC=0.771;因此,综合结合准确性、特异性、灵敏性和修斯相关系数这四个数值的计算结果来看,准确率为高。以上,仅为本发明的示意性描述,本领域技术人员应该知道,在不偏离本发明的工作原理的基础上,可以对本发明作出多种改进,这均属于本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1