一种合成奥美沙坦酯中间体杂质的方法与流程
本发明涉及医药领域,具体涉及一种合成奥美沙坦酯中间体杂质的方法。
背景技术:
奥美沙坦酯(Olmesartan Medoxomil)是由日本Sankyo(三共公司)和美国Forest Laboratories共同开发,并于2002年5月以商品名Benicar在美国上市,同年10月初在德国上市。奥美沙坦酯为前药,其代谢产物奥美沙坦是生理活性的药物,其显著特点是半衰期较长,可以在一天内有效的控制血压,因此服用比较方便,与其他的血管紧张素II受体拮抗剂类药物相比,具有剂量小、起效快、降压作用更强而持久,不良反应的发生率低等明显优点。此外,奥美沙坦对动脉硬化、心机肥厚、心力衰竭、糖尿病、肾病等均具有较好的作用。
关于奥美沙坦酯制备的专利文献比较多,比较经典的奥美沙坦酯的制备路线如图1,由于工艺中的杂质含量影响药品的质量和安全性,同时根据ICH指导原则,在API中出现的杂质含量≥0.10%都应该鉴别、合成并且表征,在制备奥美沙坦酯过程中,发现3个未知杂质,但现阶段对这些杂质的报道很少。
技术实现要素:
本发明的目的在于提供一种合成奥美沙坦酯中间体杂质的方法。
为实现上述目的,本发明采用技术方案为:
一种合成奥美沙坦酯中间体杂质的方法,奥美沙坦酯中间体杂质分别为杂质C、杂质D和杂质F,具体各自的合成过程为:
杂质C的反应式为:
按照8~15:1(g/g)质量比将75%乙酸溶液与中间体I混合,搅拌过程中升温到30~75℃,反应3~8h,待反应结束后降温20~30℃,过滤,滤液中加入与其相同体积的二氯甲烷与饮用水进行分液处理,分液后水相经二氯甲烷萃取,萃取所得有机相与分液处理的有机相合并,而后处理即得浅黄色油状物的杂质C粗品;
杂质D的反应式为:
按照5~10:1(g/g)质量比将DMF与起始物料B{4-[2-(三苯甲基四唑-5-基)苯基]苄基溴)}混合,搅拌过程中加入K2CO3,加料完毕后升温到30~75℃,反应5~12h,待反应结束后降温20~30℃,过滤,滤液中加入与其相同体积的二氯甲烷与饮用水进行分液处理,分液后水相经二氯甲烷萃取,萃取所得有机相与分液处理的有机相合并,而后处理即得油状物的杂质D粗品;其中,K2CO3与起始物料B{4-[2-(三苯甲基四唑-5-基)苯基]苄基溴)}的质量比0.5~1.5∶1(g/g);
杂质F的反应式为:
按照5~10∶1(g/g)质量比,将丙酮与中间体I混合,搅拌过程中加入KOH,加料完毕后升温到30~75℃,反应5~12h,待反应结束后降温20~30℃,浓缩体系至冷凝管不再有液滴滴下,用1M的稀盐酸调节pH=3~5,有大量固体析出,搅拌30min后过滤,干燥即可得到杂质F浅黄色粗品;其中,KOH与中间体I的质量比0.5~1.5∶1(g/g)。
所述合并有机相后依次用5%NaHCO3溶液,饱和食盐水洗涤,分液后经无水硫酸钠干燥,过滤,滤液经浓缩即得粗品,将粗品进一步纯化即得杂质C纯品。
在获得杂质C纯品时,将上述粗品中加入正庚烷和乙酸乙酯,20~30℃搅拌3~5h,过滤,滤饼干燥得纯品杂质C;其中,粗品与正庚烷的质量体积比为8~12:1(vol/g),粗品与乙酸乙酯的质量体积比为1~3∶1(vol/g)。
所述合并有机相后合并有机相,依次用5%NaHCO3溶液,饱和食盐水洗涤,分液后无水硫酸钠干燥,过滤,滤液浓缩至冷凝管不再有液滴滴下,得油状物粗品,将粗品进一步纯化即得杂质D纯品。
所述油状物粗品与硅胶混合(硅胶为200目),硅胶与粗品的质量比为1~3:1(g/g),加入二氯甲烷混匀后并浓缩,浓缩物再经柱层析分离,硅胶柱下层铺硅胶的质量与粗品质量比为5~10∶1(g/g),上层铺粗品与硅胶的混合物,经淋洗剂洗脱,收集洗脱组分,浓缩即为杂质D;其中,淋洗剂为体积比为10:1~5:1的正庚烷和乙酸乙酯混合液。
体系反应完毕后浓缩,用1M的稀盐酸调节pH=3~5,有大量固体析出,搅拌30min后过滤,干燥即可得到杂质F粗品。
在获得杂质F纯品时,将上述获得浅黄色粗品加乙酸乙酯,40~60℃搅拌1~2h,降温到20~30℃,搅拌4~6h后过滤,滤饼干燥得纯品杂质F;其中,粗品与乙酸乙酯质量体积比为4~10∶1(vol/g)。
通过1H NMR确认杂质C和杂质D以及杂质F的结构,(5)通过LC-MS图确认杂质C和杂质D的分子量。
本发明所具有的优点:
本发明制备杂质的合成方法,以便获得高纯度的对照品,同时通过优化工艺,针对性的控制这些杂质的产生,从而提高原料药的质量。同时本制备过程中只通过一步反应即可得到杂质,工艺简单;杂质D原料廉价易得,避免使用文献中提到的18冠-6催化剂,降低成本;除杂质D外,所得粗品经过重结晶即可得到粗品,操作简单。
附图说明
图1为经典的奥美沙坦酯的制备路线图。
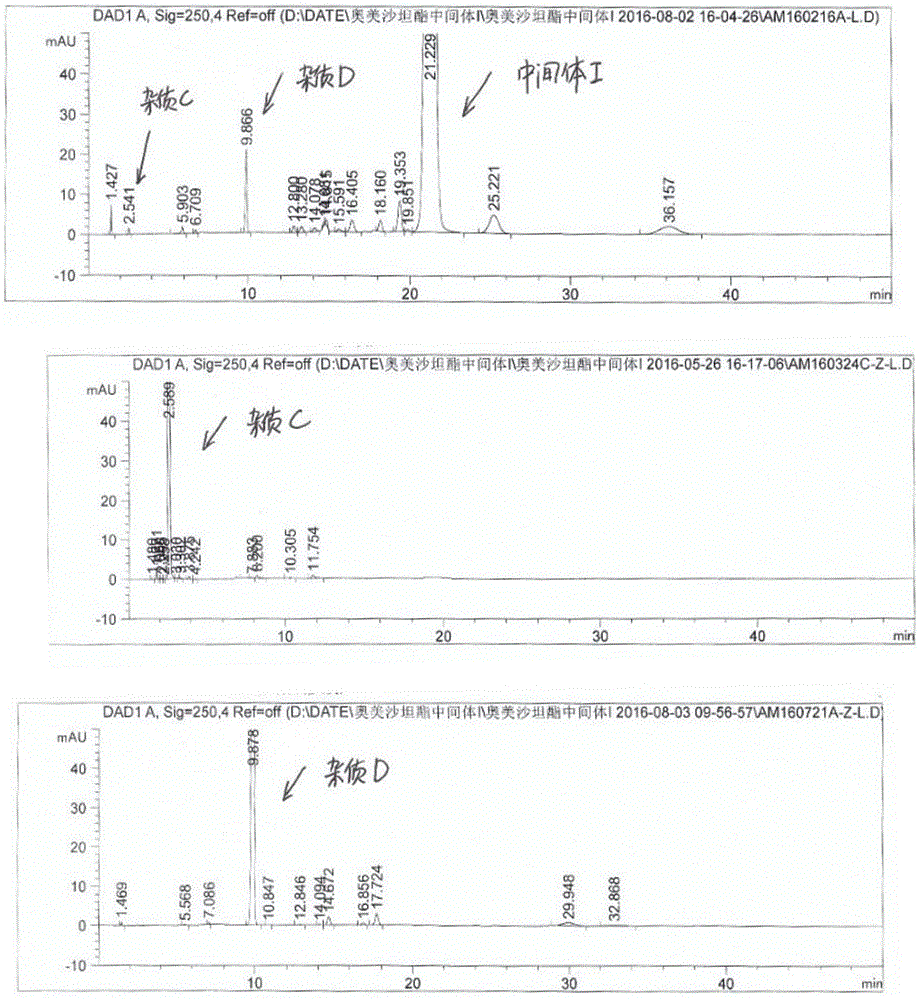
图2为本发明实施例提供的中间体I的HPLC图、杂质C的HPLC图、杂质D的HPLC图。
图3为本发明实施例提供的中间体II的HPLC图、杂质F的HPLC图。
图4为本发明实施例提供的杂质C的1H NMR与LC-MS图。
图5为本发明实施例提供的杂质D的1H NMR与LC-MS图。
图6为本发明实施例提供的杂质F的1H NMR图。
具体实施方式
通过以下实施例具体实施,对本发明的上述内容作进一步的说明。
本发明制备是(1)中间体I为原料,与乙酸反应脱保护得到杂质C,(2)起始物料B{4-[2-(三苯甲基四唑-5-基)苯基]苄基溴)}在反应体系中水解得到杂质D,(3)中间体II在反应体系中水解成羧酸得到杂质F,(4)通过1H NMR确认杂质C和杂质D以及杂质F的结构,(5)通过LC-MS图确认杂质C和杂质D的分子量。
实施例1
杂质C的合成
在装有电动搅拌器、冷凝管和温度计的500mL三颈烧瓶中,加入25.00g(34.92mmol)中间体I,250g 75%乙酸溶液,于55℃下搅拌反应3小时(TLC监测),反应停止后,降温到0~5℃,过滤,滤液加二氯甲烷150mL,饮用水150mL,分液后水相用二氯甲烷100mL萃取,分液后合并有机相,用5%NaHCO3 100mL洗涤,用饱和食盐水100mL洗涤,无水硫酸钠15.00g干燥30min,过滤有机相浓缩,得浅黄色油状液体15.23g。
油状液体中加入体积比正庚烷:乙酸乙酯=6:1混合溶剂140g,20~30℃搅拌2~5h得到10.50g杂质C(参见图2),HPLC:98.35%,收率:63.44%;
杂质D的合成
在装有电动搅拌器、冷凝管和温度计的250mL三颈烧瓶中,加入20.00g(35.88mmol)起始物料B,14.85g(107.63mmol)K2CO3和60gDMF,于50℃下搅拌反应10小时(TLC监测),反应停止后,降温到20~30℃,加二氯甲烷80mL,饮用水80mL洗涤,分液后水相用二氯甲烷60mL萃取,分液后合并有机相,用饱和食盐水80mL洗涤,,无水硫酸钠6.00g干燥30min,过滤有机相浓缩,得浅黄色油状液体10.89g。
柱层析分离:粗品加入硅胶18g(200目),加入二氯甲烷50mL溶解后减压浓缩,浓缩至不再有液滴滴下,层析柱下铺硅胶80g,体积比正庚烷:乙酸乙酯=6:1淋洗,得到6.15g杂质D(参见图2),HPLC:97.77%,收率:34.67%;
杂质F的合成
在装有电动搅拌器、冷凝管和温度计的500mL三颈烧瓶中,加入20.00g(27.93mmol)中间体I,3.13g(55.87mmol)KOH和80g丙酮,于55℃下搅拌反应3小时(TLC监测),反应停止后,降温到20~30℃,浓缩至不再有液滴滴下,用1M稀盐酸调节pH=3,有大量白色固体析出,室温搅拌1~2h,过滤,滤饼烘干后加60mL乙酸乙酯室温打浆,过滤,滤饼烘干得到11.50g杂质F(参见图3),HPLC:98.55%,收率:59.77%。
实施例2
杂质C的合成
在装有电动搅拌器、冷凝管和温度计的500mL三颈烧瓶中,加入30.00g(41.90mmol)中间体I,300g 75%乙酸溶液,于55℃下搅拌反应3小时(TLC监测),反应停止后,降温到0~5℃,过滤,滤液加二氯甲烷200mL,饮用水200mL,分液后水相用二氯甲烷150mL萃取,分液后合并有机相,用5%NaHCO3 150mL洗涤,用饱和食盐水150mL洗涤,无水硫酸钠20.00g干燥30min,过滤有机相浓缩,得浅黄色油状液体18.67g。
油状液体中加入体积比正庚烷:乙酸乙酯=6:1混合溶剂200g,20~30℃搅拌2~5h得到13.50g杂质C),HPLC:98.38%,收率:67.91%;
杂质D的合成
在装有电动搅拌器、冷凝管和温度计的250mL三颈烧瓶中,加入25.00g(44.84mmol)起始物料B,18.56g(134.54mmol)K2CO3和75gDMF,于50℃下搅拌反应10小时(TLC监测),反应停止后,降温到20~30℃,加二氯甲烷100mL,饮用水100mL洗涤,分液后水相用二氯甲烷75mL萃取,分液后合并有机相,用饱和食盐水100mL洗涤,,无水硫酸钠8.00g干燥30min,过滤有机相浓缩,得浅黄色油状液体13.66g。
柱层析分离:粗品加入硅胶20g(200目),加入二氯甲烷50mL溶解后减压浓缩,浓缩至不再有液滴滴下,层析柱下铺硅胶90g,体积比正庚烷:乙酸乙酯=6∶1淋洗,得到8.34g杂质D,HPLC:97.23%,收率:37.60%;
杂质F的合成
在装有电动搅拌器、冷凝管和温度计的500mL三颈烧瓶中,加入25.00g(34.92mmol)中间体I,3.91g(69.83mmol)KOH和100g丙酮,于55℃下搅拌反应3小时(TLC监测),反应停止后,降温到20~30℃,浓缩至不再有液滴滴下,用1M稀盐酸调节pH=3,有大量白色固体析出,室温搅拌1~2h,过滤,滤饼烘干后加80mL乙酸乙酯室温打浆,过滤,滤饼烘干得到13.50g杂质F,HPLC:98.79%,收率:56.13%。
通过杂质LC-MS(图4图5),可得出各个杂质的结构如下:
实施例3
制备得到的杂质C和杂质D与中间体I比对(参见图2),发现中间体I中确实存在杂质C和杂质D,可见中间体I中的杂质C可通过重结晶提纯除去;杂质D是由于与反应体系中的H2O发生的副反应,通过控制溶剂中的水分即可控制杂质D的产生;制备得到的杂质F与中间体II比对(参见图3),发现中间体II中确实存在杂质F,杂质F也是与溶剂的H2O发生的副反应,通过控制溶剂中的水分即可控制杂质F在中间体II的产生。
最后说明的是尽管参照较佳的实例对本方案进行详尽的说明,但不局限于此,只是用于帮助理解本发明,对于本技术领域的人员而言,在不脱离本发明原理的基础上,还可以对本发明进行若干改进,而这些改进也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!