一种通过正负电荷引入辅助病毒样颗粒自组装的方法和应用与流程
本发明涉及通过正负电荷引入改善病毒样颗粒自组装和稳定性的研究及应用,属于生物材料中的纳米载体研究领域。
背景技术:
病毒样颗粒(virus-like particle,VLP)是由病毒衣壳结构蛋白自组装而形成的空心纳米颗粒,不含病毒本身的核酸遗传物质而没有感染性。但VLP具有与天然病毒相近的结构特征,因此具有很强的免疫原性和生物学活性,适合进行相关疫苗的研发。例如,能够抵御乙型肝炎病毒(HBV)和人乳头瘤病毒(HPV)的VLP疫苗已经成功地应用于临床。此外,VLP还可作为载体平台,通过化学偶联或基因融合的方式形成嵌合型VLP,或者包裹核酸或小分子。因此,病毒样颗粒作为一种蛋白类的纳米颗粒,在免疫学、基因诊断、药物输送和材料学等领域具有广阔应用前景。例如,鼠多瘤病毒样颗粒呈二十面体,直径45nm,由72个向右歪斜排列的衣壳粒组成,而每个衣壳粒包含5个主要衣壳蛋白VP1。研究报道,结构蛋白VP1表达纯化后通常以衣壳粒(Cap)的五聚体形式存在。鼠多瘤病毒样颗粒目前通常使用两步透析法进行自组装。首先在自组装缓冲液中透析17h,再转入稳定缓冲液中透析24h,流程复杂。因而,提高病毒样颗粒的自组装效率和稳定性对其应用研究具有重大意义。
技术实现要素:
本发明目的在于提出一种通过正负电荷引入辅助病毒样颗粒自组装的方法和应用,该方法在鼠多瘤病毒样颗粒的实验验证有效。通过该方法构建鼠多瘤病毒样颗粒的两种突变体,m+VP1和m-VP1,按比例直接混合后能够自组装。紫外检测和浊点变温实验证实正负电荷引入使病毒样颗粒自组装效率和热稳定性均得到提高。
本发明的技术方案如下:
一种通过正负电荷引入辅助病毒样颗粒自组装的方法,通过构建病毒样颗粒结构蛋白的两种突变体,其一通过精氨酸或者赖氨酸等酸性氨基酸引入正电荷,另一通过谷氨酸或天冬氨酸等碱性氨基酸引入负电荷,通过正负电荷间的静电吸引提高病毒样颗粒的自组装效率和稳定性。
本发明在鼠多瘤病毒样颗粒改造和自组装强化中的应用,步骤如下:
(1)通过基因工程操作在鼠多瘤病毒样颗粒结构蛋白VP1的HI环上插入精氨酸,构建带正电的VP1突变体(m+VP1),通过插入天冬氨酸构建带负电的VP1(m-VP1),同时表达野生型VP1(wtVP1);
(2)将构建质粒转入大肠杆菌,发酵表达目标蛋白;
(3)菌液处理,获得含有目标蛋白的上清液;
(4)通过GST亲和柱分离纯化VP1,使用凝血酶切割GST标签,使用凝胶过滤色谱分离去除GST标签,获得纯化的VP1;
(5)通过引入的正负电荷辅助VLP自组装,获得稳定性好,易于自组装的VLP突变体,mVLP。
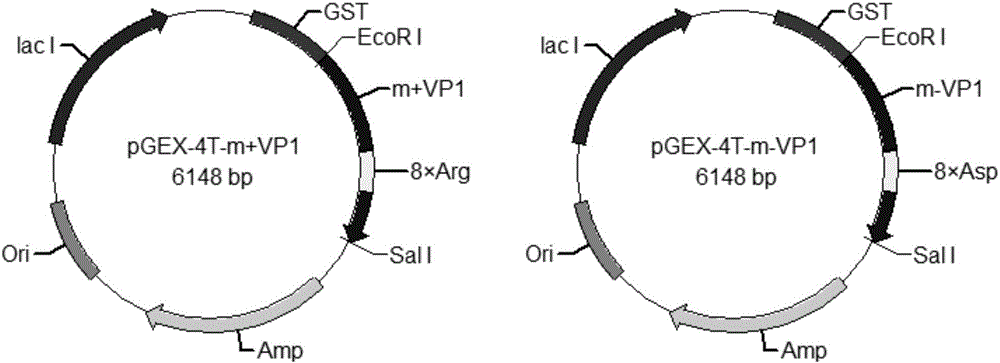
所述步骤(1)的方法是:在带有VP1基因的质粒pGEX-4T中插入外源基因序列,根据大肠杆菌密码子偏好性在VP1的HI环分别插入精氨酸密码子和天冬氨酸密码子,构建得质粒pGEX-4T-m-VP1和pGEX-4T-m+VP1。
所述步骤(2)的方法是:将构建质粒转入大肠杆菌,然后接种于氨苄氨青霉素的LB培养基培养;再接种到氨苄氨青霉素的TB培养基中,养至菌悬液OD600为0.5~0.6;加入诱导剂IPTG,转移至26℃下继续培养20~30h,诱导外源蛋白的表达。
所述步骤(3)的方法是:收集菌体,加入预冷的缓冲液重悬菌体;冰水浴超声破碎,以除去细胞碎片,获得含有目标蛋白的上清液。
所述步骤(4)的方法是:获得的含有目标蛋白的上清液通过GSTrap HP亲和柱进行分离:使用缓冲液平衡亲和柱,使用洗脱缓冲液洗脱,获得纯化的目标蛋白;使用凝血酶切除目标蛋白连接的GST标签;凝胶过滤色谱去除GST标签以分离纯化目标蛋白。
所述步骤(5)的方法是:透析法自组装或直接混合法自组装。
本发明的正负电荷引入辅助病毒样颗粒自组装的方法适用于构建表面均匀分布正负电荷位点的病毒样颗粒方面的应用。
本发明的正负电荷引入辅助病毒样颗粒自组装的方法适用于多位点蛋白自组装体方面的应用。
本发明的正负电荷引入辅助病毒样颗粒自组装的方法提高病毒样颗粒自组装效率方面的应用。
本发明的正负电荷引入辅助病毒样颗粒自组装的方法提高病毒样颗粒稳定性方面的应用。
本发明的正负电荷引入辅助病毒样颗粒自组装的方法适用于多酶固定载体方面的用途
本发明通过正负电荷引入的方法改造鼠多瘤病毒样颗粒结构蛋白,可以提高其自组装效率和稳定性。所得病毒样颗粒突变体表面均匀分布正负电荷,可以通过静电作用来固定两种带电不同的负载物,促进生物类载体开发和多酶复合物等体系的构建。
附图说明
图1:本发明构建的突变型鼠多瘤病毒样颗粒质粒图。
图2:本发明的突变型鼠多瘤病毒样颗粒的透射电镜照片。
具体实施方式
下面结合附图对本发明作详细描述,该实施例是用于解释、而不以任何方式限制本发明。
本发明的正负电荷引入辅助病毒样颗粒自组装方法应用于鼠多瘤病毒样颗粒的技术方案概述如下:
(1)通过基因工程操作在鼠多瘤病毒样颗粒结构蛋白VP1的HI环上插入精氨酸,构建带正电的VP1突变体(m+VP1),通过插入天冬氨酸构建带负电的VP1(m-VP1),同时表达野生型VP1(wtVP1);
(2)将构建质粒转入大肠杆菌,发酵表达目标蛋白;
(3)菌液处理,获得含有目标蛋白的上清液;
(4)通过GST亲和柱分离纯化VP1,使用凝血酶切割GST标签,使用凝胶过滤色谱分离去除GST标签,获得纯化的VP1;
(5)通过引入的正负电荷辅助VLP自组装,获得稳定性好,易于自组装的VLP突变体,mVLP。
具体优选各步骤如下,但不局限于此,凡是达到本发明目的的方法,都使用本发明。
所述步骤(1)的方法如下:
在带有VP1基因的质粒pGEX-4T中插入外源基因序列,根据大肠杆菌密码子偏好性在VP1的HI环分别插入精氨酸密码子和天冬氨酸密码子,构建得质粒pGEX-4T-m-VP1和pGEX-4T-m+VP1,如图1所示。
所述步骤(2)的方法如下:
将构建质粒转入大肠杆菌,然后接种于25mL含有50~100μg/mL氨苄氨青霉素的LB培养基(10g/L胰蛋白胨,5g/L酵母提取物,10g/L氯化钠)中,在37℃,170rpm条件培养10~15h;将种子液分别按1:100~1:1000的比例接种到250mL含50~100μg/mL氨苄氨青霉素的TB培养基(12g/L胰蛋白胨,24g/L酵母提取物,0.4%(v/v)甘油,2.31g/L KH2PO4,12.54g/L K2HPO4)中,在37℃,170rpm条件培养至菌悬液OD600为0.5~0.6。加入诱导剂IPTG(终浓度0.2~0.3mmol/L),转移至26℃下继续培养20~30h,诱导外源蛋白的表达;
所述步骤(3)的方法如下:
菌液在4℃,4000~5000rpm离心15~30min,收集菌体,加入预冷的L缓冲液(50mM Tris,pH 7.4,1mM EDTA,5%甘油,200mM NaCl,4mM DTT)重悬菌体,250mL菌液离心获得的菌体加60~100mL重悬液;冰水浴超声破碎,破碎条件为:200~400W,40%输出功率,工作4s,间歇6s,70~100个循环。破菌液在4℃,10000~12000rpm离心15~30min以除去细胞碎片,获得含有目标蛋白的上清液。
所述步骤(4)的方法如下:
获得的含有目标蛋白的上清液通过5mL GSTrap HP亲和柱进行分离:使用L缓冲液以0.4~0.8mL/min的流速平衡亲和柱,以0.3~0.6mL/min流速上样,之后使用洗脱缓冲液E(40mM Tris,pH 8.0,10mM还原性谷胱甘肽,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT)以0.4~0.6mL/min的流速洗脱,获得纯化的目标蛋白。
使用凝血酶切除目标蛋白连接的GST标签:将550μL纯化目标蛋白置于1.5mL的PE管中,加入0.3~0.6mg凝血酶,22℃酶切1.5~3.5h;
凝胶过滤色谱去除GST标签以分离纯化目标蛋白:使用LU缓冲液(40mM Tris,pH 8.0,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT,0.5M尿素)以0.15~0.25mL/min的流速平衡凝胶过滤柱Superdex 200;将酶切后所得溶液以同样流速上样;使用LU缓冲液洗脱,保存出峰位置的蛋白,通过电泳验证目标蛋白。
所述步骤(5)的方法如下:
透析法自组装:分离纯化所得wtVP1和m+VP1,m-VP1按比例混合,室温下置于透析袋(截留分子量8000~14000)中,先用自组装缓冲液(0.5M(NH4)2SO4,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2)透析17h,再转入稳定缓冲液(200mM NaCl,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2)中透析24h。
直接混合法自组装:将分离纯化所得wtVP1和m+VP1,m-VP1按比例混合,置于1.5mL PE管中,加入CaCl2至终浓度为1mmol/L,室温放置12~28h。
所得自组装体系滴到碳支持膜铜片,晾干后使用2%磷钨酸(pH 7.4)负染色,使用场发射透射电子显微镜观察样品形态,如图2所示。
具体说明和验证如下:
实施例1:鼠多瘤病毒样颗粒突变体的质粒构建。
在带有鼠多瘤病毒样颗粒结构蛋白VP1的基因的质粒pGEX-4T中插入外源基因序列。根据大肠杆菌E.coil BL21(DE3)密码子偏好性在VP1的Arg294和Asn295之间对应位点插入8个精氨酸密码子(CGTCGTCGTCGTCGTCGTCGTCGT),构建质粒pGEX-4T-m-VP1;插入8个天冬氨酸密码子(GATGATGATGATGATGATGATGAT),构建质粒pGEX-4T-m+VP1。
实施例2:鼠多瘤病毒样颗粒突变体的制备。
将突变质粒转入大肠杆菌BL21(DE3)中,然后接种于25mL含有100μg/mL氨苄氨青霉素的LB培养基(10g/L胰蛋白胨,5g/L酵母提取物,10g/L氯化钠)中,在37℃,170rpm条件培养12h。将种子液分别按1:1000的比例接种到250mL含100μg/mL氨苄氨青霉素的TB培养基(12g/L胰蛋白胨,24g/L酵母提取物,0.4%(v/v)甘油,2.31g/L KH2PO4,12.54g/L K2HPO4)中,在37℃,170rpm条件培养至菌悬液OD600为0.5~0.6。加入诱导剂IPTG(终浓度0.3mmol/L),转移至26℃下继续培养24h,诱导外源蛋白的表达。
菌液在4℃,4500rpm离心15min,收集菌体,加入预冷的L缓冲液(50mM Tris,pH 7.4,1mM EDTA,5%甘油,200mM NaCl,4mM DTT)重悬菌体,250mL菌液离心获得的菌体加90mL重悬液。冰水浴超声破碎,破碎条件为:300W,40%输出功率,工作4s,间歇6s,90个循环。破菌液在4℃,12000rpm离心15min以除去细胞碎片,获得含有目标蛋白的上清液。
获得的含有目标蛋白wt VP1,或m-VP1,或m+VP1的上清液通过5mL GSTrap HP亲和柱进行分离:使用L缓冲液以0.5mL/min的流速平衡亲和柱,以0.4mL/min流速上样,之后使用洗脱缓冲液E(40mM Tris,pH 8.0,10mM还原性谷胱甘肽,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT)以0.5mL/min的流速洗脱,获得纯化的目标蛋白wtVP1,或m-VP1,或m+VP1。
使用凝血酶切除目标蛋白连接的GST标签,将550μL纯化目标蛋白wt VP1,或m-VP1,或m+VP1置于1.5mL的PE管中,加入0.4mg凝血酶,22℃酶切3h;
使用LU缓冲液(40mM Tris,pH 8.0,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT,0.5M尿素)以0.2mL/min的流速平衡凝胶过滤柱Superdex 200;将酶切后所得溶液以同样流速上样;使用LU缓冲液洗脱,保存出峰位置的蛋白,通过电泳验证目标蛋白。
实施例3:鼠多瘤病毒样颗粒突变体的制备。
将突变质粒转入大肠杆菌BL21(DE3)中,然后接种于25mL含有75μg/mL氨苄氨青霉素的LB培养基(10g/L胰蛋白胨,5g/L酵母提取物,10g/L氯化钠)中,在37℃,170rpm条件培养10h。将种子液分别按1:500的比例接种到250mL含75μg/mL氨苄氨青霉素的TB培养基(12g/L胰蛋白胨,24g/L酵母提取物,0.4%(v/v)甘油,2.31g/L KH2PO4,12.54g/L K2HPO4)中,在37℃,170rpm条件培养至菌悬液OD600为0.5~0.6。加入诱导剂IPTG(终浓度0.3mmol/L),转移至26℃下继续培养30h,诱导外源蛋白的表达。
菌液在4℃,5000rpm离心20min,收集菌体,加入预冷的L缓冲液(50mM Tris,pH 7.4,1mM EDTA,5%甘油,200mM NaCl,4mM DTT)重悬菌体,250mL菌液离心获得的菌体加100mL重悬液。冰水浴超声破碎,破碎条件为:400W,40%输出功率,工作4s,间歇6s,70个循环。破菌液在4℃,11000rpm离心20min以除去细胞碎片,获得含有目标蛋白的上清液。
获得的含有目标蛋白wt VP1,或m-VP1,或m+VP1的上清液通过5mL GSTrap HP亲和柱进行分离,使用L缓冲液以0.8mL/min的流速平衡亲和柱,以0.6mL/min流速上样,之后使用洗脱缓冲液E(40mM Tris,pH 8.0,10mM还原性谷胱甘肽,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT)以0.6mL/min的流速洗脱,获得纯化的目标蛋白wtVP1,或m-VP1,或m+VP1。
将550μL纯化目标蛋白wt VP1,或m-VP1,或m+VP1置于1.5mL的PE管中,加入0.6mg凝血酶,22℃酶切1.5h;
使用LU缓冲液(40mM Tris,pH 8.0,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT,0.5M尿素)以0.25mL/min的流速平衡凝胶过滤柱Superdex 200。将酶切后所得溶液以同样流速上样。使用LU缓冲液洗脱,保存出峰位置的蛋白,通过电泳验证目标蛋白。
实施例4:鼠多瘤病毒样颗粒突变体的制备。
将突变质粒转入大肠杆菌BL21(DE3)中,然后接种于25mL含有50μg/mL氨苄氨青霉素的LB培养基(10g/L胰蛋白胨,5g/L酵母提取物,10g/L氯化钠)中,在37℃,170rpm条件培养15h。将种子液分别按1:100的比例接种到250mL含50μg/mL氨苄氨青霉素的TB培养基(12g/L胰蛋白胨,24g/L酵母提取物,0.4%(v/v)甘油,2.31g/L KH2PO4,12.54g/L K2HPO4)中,在37℃,170rpm条件培养至菌悬液OD600为0.5~0.6。加入诱导剂IPTG(终浓度0.3mmol/L),转移至26℃下继续培养20h,诱导外源蛋白的表达。
菌液在4℃,4000rpm离心30min,收集菌体,加入预冷的L缓冲液(50mM Tris,pH 7.4,1mM EDTA,5%甘油,200mM NaCl,4mM DTT)重悬菌体,250mL菌液离心获得的菌体加60mL重悬液。冰水浴超声破碎,破碎条件为:200W,40%输出功率,工作4s,间歇6s,100个循环。破菌液在4℃,10000rpm离心30min以除去细胞碎片,获得含有目标蛋白的上清液。
获得的含有目标蛋白wt VP1,或m-VP1,或m+VP1的上清液通过5mL GSTrap HP亲和柱进行分离,使用L缓冲液以0.4mL/min的流速平衡亲和柱,以0.3mL/min流速上样,之后使用洗脱缓冲液E(40mM Tris,pH 8.0,10mM还原性谷胱甘肽,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT)以0.4mL/min的流速洗脱,获得纯化的目标蛋白wtVP1,或m-VP1,或m+VP1。
将550μL纯化目标蛋白wt VP1,或m-VP1,或m+VP1置于1.5mL的PE管中,加入0.3mg凝血酶,22℃酶切3.5h;
使用LU缓冲液(40mM Tris,pH 8.0,200mM NaCl,1mM EDTA,5%(v/v)甘油,5mM DTT,0.5M尿素)以0.15mL/min的流速平衡凝胶过滤柱Superdex 200。将酶切后所得溶液以同样流速上样。使用LU缓冲液洗脱,保存出峰位置的蛋白,通过电泳验证目标蛋白。
实施例5:正负电荷引入辅助病毒样颗粒自组装。
分离纯化所得wtVP1和m+VP1,m-VP1等比例混合,室温下置于透析袋(截留分子量8000~14000)中,先用自组装缓冲液(0.5M(NH4)2SO4,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2)透析17h,再转入稳定缓冲液(200mM NaCl,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2)中透析24h。所得自组装体系滴到碳支持膜铜片,晾干后使用2%磷钨酸(pH 7.4)负染色,使用场发射透射电子显微镜观察样品形态,如图2所示。
自组装体的电镜结果显示,m+VP1:m-VP1等比例混合体系形成规则颗粒,如图2所示,而同等条件的野生型体系没有任何变化,说明正负电荷的引入能够很好促进病毒样颗粒的自组装。
表达纯化所得VP1蛋白,包括wtVP1,m-VP1和m+VP1,均使用圆二色测定其二级结构,扫描光谱范围为190~260nm。
结果表明,m-VP1和m+VP1与wtVP1具有相同二级结构,说明带正电或带负电序列的插入并不影响结构蛋白的二级结构。
实施例6:病毒样颗粒突变体的自组装效率检测。
牛血清白蛋白(BSA)通过去离子水溶解获得浓度为50mg/mL的溶液。在96微量孔板上,每孔滴加300μL BSA溶液,于室温下放置2h,封闭微孔中的结合位点。随后每次使用200μL的自组装缓冲液(0.5M(NH4)2SO4,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2)润洗微孔3次。
100μL wtVP1,m+VP1,m-VP1的等比例混合物(蛋白终浓度为0.15mg/mL)加入已封闭结合位点的微孔中,再各加入200μL自组装缓冲液(0.5M(NH4)2SO4,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2),混匀后使用酶标仪(Infinite M200,TECAN)检测350nm的紫外吸收值,检测持续17h。
结果表明,随着自组装时间推移,突变型VP1等比例混合物的OD350增加速率快于野生型VP1,说明突变型结构蛋白的自组装效率高于野生型。
实施例7:突变型病毒样颗粒的热稳定性检测。
利用紫外可见分光光度计检测wtVLP和mVLP的热稳定性。蛋白样品分别溶解于稳定缓冲液(200mM NaCl,20mM Tris,pH 7.4,5%(v/v)甘油,1mM CaCl2)中,样品终浓度为0.05mg/mL,检测前使用0.22μm的滤膜过滤蛋白样品。将1mL蛋白样品置于1cm×1cm比色皿中,检测逐渐递增温度时样品在350nm处的紫外吸收,测温范围30~80℃。每个样品测试三次,取平均值。
结果表明wtVLP的Tm为52℃,而mVLP的Tm为59.2℃,表明mVLP的热稳定性高于野生型。
综上,实验结果说明正负电荷的引入可以提高病毒样颗粒的自组装效率和热稳定性,该方法是一种改善病毒样颗粒自组装和稳定性的有效方法。
本发明提出一种通过正负电荷引入辅助病毒样颗粒自组装的方法和应用。无透析组装、自组装过程紫外吸光值实时检测、紫外浊点检测等实验全面考察病毒样颗粒的自组装效率和热稳定性,已通过实施例进行描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法进行改动或适当变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替代和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本发明精神、范围和内容中。
- 还没有人留言评论。精彩留言会获得点赞!