一种用于制备可异体移植T细胞的基因编辑系统的制作方法
本发明涉及基因靶向编辑领域,尤其涉及一种利用基因编辑技术制备可异体移植t细胞技术,具体的为一种用于制备可异体移植t细胞的基因编辑系统。
背景技术:
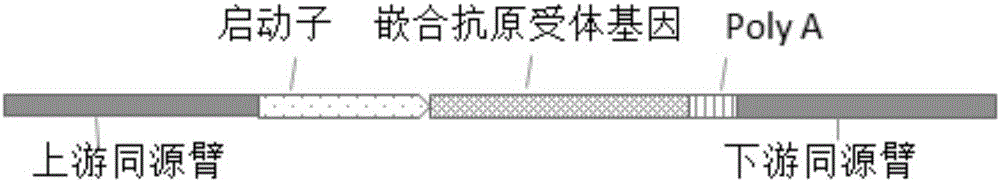
:近年来,利用免疫疗法来治疗肿瘤,受到广泛的关注。在众多的免疫疗法中,基于嵌合抗原受体t细胞(chimericantigenreceptortcells,car-t)疗法,效果最为显著和成熟。目前已经有多例临床试验证明,该技术可以治愈多种肿瘤,例如b细胞肿瘤。其基本原理是利用基因工程的方法,将能识特异识别肿瘤细胞的受体基因,利用整合型病毒作为载体,将该受体基因整合到患者t细胞基因组中,从而使改造后的t细胞具有特异性识别和杀伤肿瘤细胞的目的,从而达到治疗肿瘤的效果。另一方面,该技术还存在一些急需解决的障碍,首先,该技术只能针对病人自体t细胞进行编辑,大大提高了其治疗成本及延长治疗周期(利用病人自身t细胞制备car-t细胞,一般耗时长);第二,由于t细胞表面不仅表达car还有自身的tcr受体表达,研究发现tcr受体会影响car受体的功能,有临床研究表面,可对神经系统造成伤害,形成脑水肿。为解决以上car-t治疗技术的问题,多个研究小组利用基因编辑技术(zfn,talen)针对tcr受体敲除,取得了显著效果。有研究报道,目前利用talen技术敲除tcr受体来制备mcar-t技术(mniversaicar-t),该技术一方面将tcr受体通过talen定点敲除,另一方面通过同源重组技术将car基因定点整合到tcr基因位点。该技术不仅有效的避免了异体移植导致的移植物抗宿主反应gvhd(ghostversμshostdisease),使得car-t细胞的制备摆脱个性化的缺陷而进入到标准化和规模化的时代,同时还避免了tcr基因对car受体功能的干扰。此外,由于car基因定点整合到tcr位点上,避免了现行car-t制备中car基因随机整合基因组所导致的潜在癌变风险。目前该技术已经获得了fda的临床试验批文,而且利用该技术成功为两患者进行了治疗。然而由于zfn或者talen技术属于第一,二代基因编辑技术,在应用方面存在很大的局限性,例如构建流程复杂、周期长、成本高等。近几年基因编辑技术突破性的发展,利用现在的编辑手段,可以将目的基因进行精准编辑。尤其是第三代基因编辑技术crispr具有明显的优势,例如构建简单,效率高,成本低等。crispr系统均包含以下几个部分:1)pam位点,位于靶序列的下游,只有几个nt(例如,spcas9/ngg;sacas9/nngrrt),根据此位点来选取靶序列;2)靶序列(sgrna),识别并与切割位点结合的序列,一般在20nt左右;3)tracrna,连接于靶序列之后的一段回文rna序列;4)cas9内切酶,具有独立酶活性,cas9含有在氨基末端的rμvc和蛋白质中部的hnh2个独特的活性位点,cas9中的hnh活性位点剪切sgrna的互补dna链,rμvc活性位点剪切非互补链。其工作原理为:sgrna、tracrrna和cas9组成复合体,识别并结合于sgrna互补的序列,然后解开dna双链,形成r-loop,使sgrna与互补链杂交,另一条链保持游离的单链状态,然后由cas9中的hnh活性位点剪切sgrna的互补dna链,rμvc活性位点剪切非互补链,最终引入dna双链断裂(dsb)。crispr技术目前已经广泛应用于多个领域,例如动植物育种,疾病治疗,以及基础科研等领域。crispr-sacas9系统,是一种来自金黄色葡萄球菌的cas9酶。其在哺乳动物细胞中的基因编辑效率高,而且其基因比较小(3.1kb)。因此,该系统能够利用多种转染系统转染,例如电转化、腺相关病毒等。另外由于sacas9系统pam序列较长(nngrrt),且靶点识别序列在21-23nt,导致该系统具有靶向性高、脱靶效率低等优点。鉴于现有car-t技术还有待进一步完善和优化,以及crispr/sacas9技术在基因精准编辑上等高效性和安全性,本发明将二者有机结合,用crispr/sacas9技术敲除人tcr基因,制备可异体移植的通用型t细胞,用以改善和优化car-t制备技术。crispr/cas9介导的定点同源重组必备两个条件:一是crispr/cas9,含有效切割目的基因的grna;二是同源重组供体(图1),一般含上、下游同源臂,以及在两同源臂之间的需要定点整合的外源dna序列(该外源序列可以含启动子或不含启动子,如不含启动子,则必须插入外显子序列并保持插入后读码框正确)。当宿主的基因组dna被grna识别并引导cas9定点切割,同源重组供体存在下,宿主细胞倾向于通过同源重组的方式修复基因组dna,从而在基因组dna特定位点引入了外源dna序列(图2)。经我们研究发现,同源臂序列的长度,以及外源dna序列的长度,对重组效率都有极大影响。目前我们确定最优的同源臂长度在800bp以内(含800bp),而外源dna序列长度以5kb以内为佳。采用crispr/cas9介导的定点同源重组技术,在敲除人tcr基因的同时,在tcr基因特定位点(cas9切割位点),通过同源重组技术插入嵌合抗原受体(car)基因,从而达到tcr敲除和car表达的双重效果。利用该技术可以制备特异性攻击肿瘤组织的通用型t细胞,这种通用型t细胞具有以下几方面的优点:1)敲除了tcr基因,避免了异体移植导致的移植物抗宿主反应,所以利用该技术可以实现t细胞的异体移植,实现car-t的标准化和规模化生产,降低成本并缩短生产周期;2)tcr基因敲除减低tcr对细胞表面car功能的影响,降低临床安全风险;3)car定点插入tcr位点,达到tcr敲除和car表达的双重效果,避免现有技术中心car随机插入带来的癌变风险。技术实现要素:本发明的目的在于,提供一种能够异体移植的通用型t细胞制备技术,该技术制备的t细胞具备以下优点:第一,能够异体移植,使用于制备car-t的t细胞成为一种储备型生物药品,便于大规模、批量化、标准化生产;第二,提高car受体的功能活性,避免了tcr受体的干扰。同时,利用crispr/cas9介导的tcr基因位点定点同源重组技术,该技术制备的t细胞具备以下优点:第一,tcr基因敲除,可以异体移植,规模化生产;第二,避免tcr受体干扰car受体的功能;第三,tcr基因位点定点插入外源基因,可实现car基因定点整合到tcr基因位点,避免异体移植导致的移植物抗宿主反应。本发明提供一种用于制备可异体移植t细胞的基因编辑系统,该系统包括在crispr/sacas9特异性敲除人tcr基因中用于特异性靶向tcr基因的sgrna,以及5′端和3′端带有tcr基因同源臂的嵌合抗原受体基因。该系统通过六型腺相关病毒(aav6)或慢病毒或杆状病毒为载体转染细胞。本发明的技术方案如下:一种用于制备可异体移植t细胞的基因编辑系统,该系统包括在crispr/cas9特异性敲除人tcr基因中用于特异性靶向tcr基因的sgrna,所述sgrna在tcr基因上的靶序列符合5′-g(21n)nngrrt-3或者5′-arrcnn(21n)c-3′的序列排列规则,所述系统还包括5′端和3′端带有tcr基因同源臂的嵌合抗原受体基因。一种用于制备可异体移植t细胞的基因编辑系统,该系统包括在crispr/cas9特异性敲除人tcr基因中用于特异性靶向tcr基因的sgrna,所述sgrna在tcr基因上的靶序列符合5′-g(21n)nngrrt-3或者5′-arrcnn(21n)c-3′的序列排列规则,所述靶向敲除tcr基因的靶序列以病毒为转染载体,所述系统还包括5′端和3′端带有tcr基因同源臂的嵌合抗原受体基因,其可以通过所述的病毒为载体,使用时与含有tcr基因的靶序列的病毒共同转染细胞。一种用于制备可异体移植t细胞的基因编辑系统,该系统包括在crispr/cas9特异性敲除人tcr基因中用于特异性靶向tcr基因的sgrna,所述sgrna在tcr基因上的靶序列符合5′-g(21n)nngrrt-3或者5′-arrcnn(21n)c-3′的序列排列规则,所述系统还包括5′端和3′端带有tcr基因同源臂的嵌合抗原受体基因,使用时把tcr基因的靶序列、cas9基因、5′端和3′端都带有tcr基因同源臂的嵌合抗原受体基因,三者一起包装到杆状病毒中,以杆状病毒为载体转染细胞。优选的,以所述人tcr基因α链和β链恒定区作为靶标,通过靶向tcr基因α链和β链的sgrna筛选,由此获得了在crispr-cas9特异性敲除人tcr基因中用于特异性靶向tcr基因的sgrna,所述人tcr基因α链和β链恒定区基因号为nc_000014.9;nc_000007.14。优选的,以所述人tcr基因α链和β链恒定区作为靶标,依据sacas9f.annranetal.2015编辑规则筛选出了多个靶序列,其中21n的靶序列如seqidno:1~seqidno:30中任意一条序列。优选的,所述sgrna在tcr基因上的靶序列符合5′-g(20n)ngg-3′或者5′-ccn(20n)c-3′的序列排列规则,并满足seqidno:37~seqidno:62中任意一条序列。优选的,所述靶向敲除tcr基因的靶序列配对组合为seqidno:3和seqidno:4,seqidno:3和seqidno:8,seqidno:4和seqidno:8,seqidno:3和seqidno:13,seqidno:3和seqidno:14,seqidno:3和seqidno:17)以及多个靶序列(≥3)组合形成的crispr/sacas9系统。优选的,所述靶向敲除tcr基因的靶序列以病毒为转染载体,所述病毒转染载体为腺相关病毒、慢病毒、腺病毒、杆状病毒、逆转录病毒中的一种,所述腺相关病毒包括各种血清型腺相关病毒以及其突变型,所述慢病毒包括整合型以及非整合型。优选的,所述靶向敲除tcr基因的靶序列为dna或相对应的rna。优选的,所述tcr基因包含α链和β链,所述基因改造具体方法为在tcr的α链和β链的一种或两种的cds序列或恒定区域的相应编码基因的外显子区,利用crispr/cas9进行定点切割,从而使tcr基因失活,达到t细胞表面无tcr受体的目的,编辑后的t细胞在有异体移植过程中避免移植物抗宿主反应。优选的,所述靶向tcr基因α链和β链的sgrna筛选包括如下步骤:步骤1)构建sgrna的寡核苷酸双链:s1:根据选择的sgrna,通过化学合成法合成寡核苷酸链,格式如下:正义链:5′-g(21n)nngrrt-3′或者5′-arrcnn(21n)c-3′,引物不合pam,反义链:与正义链互补,分别在正义链引物的5′端加上cacc,在反义链引物5′端加上aaac,以形成bbsi酶切位点的粘性末端;其中,21n代表靶序列的21个碱基序列;s2:把正义链引物和反义链引物经过退火,形成带有bbsi酶切位点的粘性末端的双链核苷酸小片段;步骤2)crispr-cas9载体构建;步骤3)细胞系转染;步骤4)突变效率检测。本发明还提供了一种应用,应用于上述所述的用于制备可异体移植t细胞的基因编辑系统,其特征在于,用外周血或脐带血来源的t细胞,通过上述用于制备可异体移植t细胞的基因编辑系统,制备成一种可异体移植的t细胞。附图说明图1a为本发明中同源重组供体(含启动子)的结构图;图1b为本发明中同源重组供体(不含启动子)的结构图;图2为本发明中基因重组示例图;图3为本发明中px601的结构图;图4为本发明中px458的结构图;图5为本发明中同源重组供体质粒pdonor-mcmv-egfp的结构图;图6a\6b\6c\6d\6e为本发明实施例中以dl2000为参照的电泳结果图;图7a\7b\7c为本发明实施例中以dl2000为参照的电泳结果图;图8a\8b为本发明实施例中以dl2000为参照的cr产物的长度的电泳结果图;图9为本发明实施例中t7e1的酶切和电泳结果图;图10为本发明实施例中pdonor-mcmv-egfp-上下游同源臂的结构图;图11为本发明实施例中seqidno:71的结构图。具体实施方式为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。以下实施例中使用到的实验材料包含:商品化的crispr-cas9载体,如px601(参照图3),px458(参照图4);hek293t细胞;大肠杆菌感受态细胞top10;同源重组供体质粒pdonor-mcmv-egfp(参照图5)。实施例1grna初步筛选1、针对tcra基因的grna准备(1)根据tcra基因的序列(包括包含α链和β链恒定区)设计的grna序列:crispr/spcas9系统的grna对应的靶序列如下表所示:表1针对tcra基因的crispr/spcas9系统的grna对应的靶序列。crispr/sacas9系统的grna对应的靶序列如下表所示:表2针对tcra基因的crispr/sacas9系统的grna对应的靶序列。(2)分别合成grna对应的靶序列的正义链和反义链(正义链的5′-端加cacc,若正义链5′-端第一个核苷酸不是鸟嘌呤g,则在正义链的5′-端加caccg;在反义链的5′-端加aaac,若正义链5′-端第一个核苷酸不是鸟嘌呤g,则在反义链的3′-端加c);(3)将上述grna正义链和反义链溶解成100μm的母液,各取1μl加入到98μlddh2o混合稀释到1μm,90℃处理30s后,移至室温冷却完全退火。反应体系如下:2、载体准备(1)将100μl的top1o-px601和top10-px458菌液分别加入100mllb液体培养基(amp)中,恒温摇床37℃200rpm摇菌12-16h;(2)分别提取px601质粒和px458质粒,并测定质粒浓度;(3)采用bsal对px601进行酶切,采用bbsi对px458进行酶切,37℃酶切1h,反应体系如下:(4)1%琼脂糖凝胶回收经酶切的质粒px601和px458,并测定回收酶切质粒的浓度,-20℃保存备用。3、连接转化(1)将表1对应的退火后的grna与酶切纯化过的px458载体进行连接反应;将表2对应的退火后的grna与酶切纯化过的px601载体进行连接反应。25℃连接20min,反应体系如下:(2)将上述连接产物各5μl分别加入两管20μl的top10感受态细胞中,用移液器轻轻吹打混匀,冰浴30min。42℃热击45s,迅速将离心管转移到冰浴中,冰上静置5min。向每个离心管中加入230μl无菌的lb液体培养基(不含抗生素),混匀后置于恒温摇床37℃200rpm振荡培养45min使菌体复苏。(3)将复苏后的感受态细胞直接倒至lb固体平板(amp抗性)上,晃动平板使菌液涂布均匀,超净工作台吹干平板,将平板倒置于恒温培养箱37℃静置培养12-16h。(4)从上述平板上分别挑取单菌落接种到500μllb液体培养基(amp)中扩大培养4h。(5)使用正向测序引物seqidno:63(5′-atttttgtgatgctcgtcag-3′)对px458的连接产物转化的菌液进行测序;使用正向测序引物seqidno:64(5′-ttccttgaccctggaaggtg-3′)对px601的连接产物转化的菌液进行测序;(6)将上述测序正确的菌种扩大培养后提取质粒并测定质粒浓度后至-20℃保存备用。4、细胞转染(1)hek293t细胞铺板(2)采用lipofectamine3000试剂盒将上述3(6)中提取的质粒分别转染hek293t细胞;(3)转染后的细胞培养48小时,收集细胞。5、t7e1酶切分析突变效率(1)将上述4(3)收集的细胞提取细胞基因组,并检测基因组浓度;(2)分别在grna结合位点上下游设计pcr检测引物,见表1和表2;(3)使用高保真pcr试剂盒分别扩增带有靶位点的目的片段,pcr反应条件:95℃3min→95℃15s→55℃15s→72℃30s→72℃5min,35cycles。(4)使用产物纯化试剂盒回收pcr产物,并测定产物浓度;(5)将上述纯化的pcr产物退火处理,即先加热至95℃,保温10min,然后以每30s下降2~3℃的速度降温至室温;:(6)上述每管退火产物分别加入t7核酸内切酶1(t7e1),设置mock组(未转化的细胞)和空白对照(ck)组(不加t7e1,用ddh2o代替),37℃酶切1h。(7)以dl2000(takara)为参照,用2%琼脂糖凝胶电泳检测酶切效果,电泳结果见图6和图7。图6为crispr/sacas9系统对tcra基因的grna筛选,图7为crispr/spcas9系统对tcra基因的grna筛选。(a)(b)(c)分别为以seqidno:31和seqidno:32,seqidno:33和seqidno:34,seqidno:35和seqidno:36为检测引物的grna对tcra基因的突变效率检测,泳道编号对应grna靶序列编号。根据t7e1的酶切原理,突变效率高与酶切效率成正比,图中编号带下划线的泳道酶切效率较高,即所对应的grna对tcra基因的突变效率较高。(8)在上述t7e1酶切电泳图中突变效率较高的pcr产物中,随机挑取4个,给pcr产物3′-末端加一个腺嘌呤(a),然后用1%琼脂糖凝胶电泳后分别切胶回收目的片段,并测定回收产物浓度;(9)将上述4份回收产物分别与pmdtm18-t载体进行连接反应,16℃反应30min;(10)将上述4份连接产物分别按3(2)-(3)方法转化大肠杆菌感受态细胞top10,并分别涂布lb平板(amp);(11)从上述4个平板上分别随机挑取10-50个单菌落,进行sanger测序;(12)上述测序结果统计如下表3所示。由测序结果可知:表3sanger测序比较突变效率。注:有效测序菌落总数指,随机挑取进行测序的菌落总数,减去发生载体自连的菌落数。实施例2针对tcra基因的grna组合筛选将实施例1中经过t7e1酶切和琼脂糖电泳分析,筛选出来的突变效率较高的grna,组合起来对tcra基因进行突变操作,具体步骤如下:1、grna组合如下表所示:表4针对tcra基因的crispr/sacas9系统的grna组合对应的靶序列:将实施例1中3(6)制备的质粒,按照上述组合方式,共同转染hek293t细胞,转染方式参照实施例1的4(1)~4(3);转染后48小时,按照实施例1中5(1)~5(4)的方法扩增并纯化含有靶位点的目的片段;以dl2000(takara)为参照,用2%琼脂糖凝胶电泳检测上述pcr产物的长度。结果如图8所示(a)(b)分别为以seqidno:31和seqidno:32,seqidno:31和seqidno:34为检测引物的grna靶序列组合对tcra基因的突变效率检测,泳道编号对应grna靶序列组合的编号。如表4标识,共转的grna其靶序列之间相距一定的距离,理论上双grna共同作用会在tcra基因上形成片段缺失,通过pcr产物电泳,会形成两条大小不同的条带,图8结果与理论相符,说明本方案所选择的grna组合能有效突变tcra基因。对上述pcr产物片段大小无明显变化的grna组合,按照实施例1中5(5)~5(7)的方法进行t7e1的酶切和电泳检测。结果见图9所示:seqidno:3和seqidno:4靶序列之间相距51bp,其grna组合共同突变tcra基因后其pcr产物未形成两条独立条带,但对其通过t7e1酶切进一步验证发现具有较高的突变效率。本方案所选择的grna其靶序列之间的距离覆盖了51-2322bp,同理可证,在实施例1中筛选出来的突变效率较高的靶序列中,选取靶序列相距51-2322bp之间的两个grna组合,均能有效突变tcra基因。对上述组合中随机挑取4组grna组合,按照实施例1中5(8)~5(11)的方法进行sanger测序分析。结果如下表5所示:表5sanger测序比较grna组合突变效率实施例3crispr/cas9介导的tcra基因定点同源重组将实施例1或者实施例2中筛选出来突变效率较高的grna或者grna组合,用于crispr/cas9介导的,在tcra基因特定位点(cas9切割位点),整合插入一段外源dna序列。以下实施例以grna靶序列seqidno:3为例,同理可证其他grna或者grna组合的实施效果。1、同源重组供体载体构建(1)以hek293t细胞基因组为模板,以seqidno:65和seqidno:66为引物扩增上游同源臂,以seqidno:67和seqidno:68为引物增下游同源臂,pcr产物用1%琼脂糖凝胶电泳检测后切胶回收目的片段;表6tcra基因靶位点seqidno:3上下游同源臂扩增引物65、66、67、68引物类型引物编号引物序列上游同源臂扩增引物-fseqidno:65gttctagtggttggctacgtatgctcaaggccttatatcgag上游同源臂扩增引物-rseqidno:66gtcgacctgtgggacaagaggatcag下游同源臂扩增引物-fseqidno:67actagttctagagcggccgcctgcctattcaccgattttga下游同源臂扩增引物-rseqidno:68actgcaggctctagattcgaatgcgtgagactgacttagtg(2)提取pdonor-mcmv-egfp质粒,用snabi和sali-hf酶切pdonor-mcmv-egfp质粒,37℃酶切1h,80℃失活20min;(3)酶切后的pdonor-mcmv-egfp和上游同源臂pcr产物进行同源重组,37℃重组30min,冰浴5min;(4)5μl上述重组产物和20μltop10大肠杆菌感受态混合后冰浴30min,42℃热击60s,冰上静置5min后加入230mllb培养基(不加抗生素),37℃摇床45min,涂板,37℃培养过夜;(5)从上述平板上分别挑取单菌落接种到500μllb液体培养基(amp)中扩大培养4h后测序;(6)扩大培养测序正确(上游同源臂重组到pdonor-mcmv-egfp载体上)的单菌落并提取质粒,用noti-hf,37℃酶切1h,然后加入bstbi,65℃酶切1h,酶切后纯化回收;(7)上述酶切产物与下游同源臂pcr产物进行同源重组,37℃重组30min,冰浴5min;(8)按本实施例4~5的方法转化top10大肠杆菌感受态、挑取单菌落,测序鉴定,序列正确的质粒即为同源重组供体载体pdonor-mcmv-egfp-上下游同源臂(图10),对其扩大培养和保存,。2、tcra基因定点同源重组(1)将实施例1中3(6)中提取的seqidno:3对应的质粒,与上述同源重组供体质粒共同转染hek293t细胞,转染方式参照实施例1的4(1)~4(3);(2)转化后的细胞培养48小时,收集细胞提取基因组,并测定基因组浓度;(3)以上述基因组dna为模板,在上游同源臂的5′端设计上游pcr引物(seqidno:69∶5′-ctgtggctctgcatgactcactag-3′),在外源dna序列mcmv-egfp中间设计下游引物(seqidno:70∶5′-atcgccttcttgacgagttcttctgag-3′),进行pcr扩增,pcr产物进行sanger测序。测序结果见seqidno:71,结果分析见图11所示:5′端126bp为tcra基因的序列,127-731bp为上游同源臂,732-1266bp为同源重组供体质粒的外源dna序列的一部分,含mcmv启动子和部分egfp基因序列。该结果说明,同源重组供体质粒的外源dna序列已成功整合到tcra基因的特定位点。按照本实施例的方法,同理可将嵌合抗原受体(car)基因定点同源重组到tcr的特定位点。seqidno:71:如下:attaaatagatgtttatatggagaagctctcatttctttctcagaagagcctggctaggaaggtggatgaggcaccatattcattttgcaggtgaaattcctgagatgtaaggagctgctgtgacttgctcaaggccttatatcgagtaaacggtagcgctggggcttagacgcaggtgttctgatttatagttcaaaacctctatcaatgagagagcaatctcctggtaatgtgatagatttcccaacttaatgccaacataccataaacctcccattctgctaatgcccagcctaagttggggagaccactccagattccaagatgtacagtttgctttgctgggcctttttcccatgcctgcctttactctgccagagttatattgctggggttttgaagaagatcctattaaataaaagaataagcagtattattaagtagccctgcatttcaggtttccttgagtggcaggccaggcctggccgtgaacgttcactgaaatcatggcctcttggccaagattgatagcttgtgcctgtccctgagtcccagtccatcacgagcagctggtttctaagatgctatttcccgtataaagcatgagaccgtgacttgccagccccacagagccccgcccttgtccatcactggcatctggactccagcctgggttggggcaaagagggaaatgagatcatgtcctaaccctgatcctcttgtcccacaggtcgacggtaggcgtgtacggtgggaggtctatataagcagagctggtttagtgaaccgtcagatcaccggtcgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatsequencelisting<110>广东赤萌医疗科技有限公司<120>一种用于制备可异体移植t细胞的基因编辑系统<130>1<160>1<210>1<211>1266<212>dna<213>人工序列<400>1attaaatagatgtttatatggagaagctctcatttctttctcagaagagc50ctggctaggaaggtggatgaggcaccatattcattttgcaggtgaaattc100ctgagatgtaaggagctgctgtgacttgctcaaggccttatatcgagtaa150acggtagcgctggggcttagacgcaggtgttctgatttatagttcaaaac200ctctatcaatgagagagcaatctcctggtaatgtgatagatttcccaact250taatgccaacataccataaacctcccattctgctaatgcccagcctaagt300tggggagaccactccagattccaagatgtacagtttgctttgctgggcct350ttttcccatgcctgcctttactctgccagagttatattgctggggttttg400aagaagatcctattaaataaaagaataagcagtattattaagtagccctg450catttcaggtttccttgagtggcaggccaggcctggccgtgaacgttcac500tgaaatcatggcctcttggccaagattgatagcttgtgcctgtccctgag550tcccagtccatcacgagcagctggtttctaagatgctatttcccgtataa600agcatgagaccgtgacttgccagccccacagagccccgcccttgtccatc650actggcatctggactccagcctgggttggggcaaagagggaaatgagatc700atgtcctaaccctgatcctcttgtcccacaggtcgacggtaggcgtgtac750ggtgggaggtctatataagcagagctggtttagtgaaccgtcagatcacc800ggtcgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgc850ccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtg900tccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagtt950catctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgacca1000ccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaag1050cagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcg1100caccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtga1150agttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgac1200ttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaa1250cagccacaacgtctat1266当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1