编辑DNA甲基化的方法与流程
本申请要求2016年8月19日提交的美国临时申请序列号62/377,520的权益,该美国临时申请的内容在此以引用的方式整体并入本文。
政府支持
本发明是在政府支持下根据国家卫生研究院(nationalinstitutesofhealth)授予的基金号hd045022作出的。政府享有本发明的某些权利。
背景技术:
5-胞嘧啶处的哺乳动物dna甲基化在许多生物过程中发挥关键的作用,包括基因组印记、细胞命运决定、染色质结构组织、细胞身份的维持以及基因表达的调节(bird,2002;cedar和bergman,2012;jaenisch和bird,2003;smith和meissner,2013)。遗传研究已经揭示,dna甲基化对于哺乳动物发育和对环境信号的适应来说是必要的(jaenisch和bird,2003;li等,1992;smith和meissner,2013)。异常的dna甲基化已经在癌症和神经病症中被观测到(laird和jaenisch,1996;robertson,2005)。由于测序技术的进步,因此许多类型的人类和小鼠细胞和组织的单核苷酸分辨率甲基化图谱已经被描绘出(lister等,2009;schultz等,2015)。重要的是,这些图谱已经允许在正常发育(lister等,2013)以及疾病(dejager等,2014;doi等,2009;landau等,2014)的不同阶段期间以碱基对分辨率鉴定出差异甲基化区域(dmr)。然而,由于缺乏使得能够以靶向方式有效编辑dna甲基化的适当分子工具,因此对这些dmr的功能意义的研究仍然是一项挑战。
技术实现要素:
哺乳动物dna甲基化是在许多生物过程中协调基因表达网络的表观遗传机制。然而,对特定甲基化事件的功能的研究仍然具有挑战性。已经证实,tet1或dnmt3a与无催化活性的cas9(dcas9)的融合使得能够进行靶向dna甲基化编辑。使dcas9-tet1融合蛋白或dcas9-dnmt3a融合蛋白靶向到甲基化的启动子序列或非甲基化的启动子序列分别引起内源性报告基因的激活或沉默。dcas9-tet1对bdnf启动子iv或myod远端增强子进行的靶向脱甲基化分别诱导有丝分裂后神经元中的bdnf表达或激活myod,从而促进成纤维细胞重编程为成肌细胞。dcas9-dnmt3a对ctcf环锚定位点进行的靶向从头甲基化阻断ctcf结合并且干扰dna成环,从而引起相邻环中的基因表达改变。最终,证实这些工具可以在小鼠中编辑dna甲基化,这表明了它们对于表观遗传调节的功能研究的广泛效用。这些工具将可用于深入了解dna甲基化在诸如基因表达、细胞命运决定以及高阶染色质结构的组织的不同生物过程中的功能意义。此外,这些工具将可用于构建筛选平台以当与不同的sgrna文库组合时鉴定功能上指定的差异甲基化区域(dmr)以及产生转基因小鼠以在体内研究特定dna甲基化事件。
本文公开了修饰细胞中的一个或多个基因组序列的方法,所述方法包括向所述细胞中引入与具有甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶;以及一个或多个指导序列,从而修饰所述细胞中的一个或多个基因组序列。
本文还公开了修饰细胞中的一个或多个基因组序列的方法,所述方法包括向所述细胞中引入与具有脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶;以及一个或多个指导序列,从而修饰所述细胞中的一个或多个基因组序列。
本文还公开了调节细胞中一个或多个基因组序列的甲基化的方法,所述方法包括向所述细胞中引入与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶;以及指导序列或编码指导序列的核酸,从而调节细胞中一个或多个基因组序列的甲基化。
在某些方面,所述基因组序列包括差异甲基化区域、增强子(例如myod的增强子)、启动子(例如bdnf启动子)或ctcf结合位点。在一些方面,所述效应子结构域包括tet1。在其它方面,所述效应子结构域包括dnmt3a。
在一些方面,所述无催化活性的位点特异性核酸酶是无催化活性的cas蛋白(例如cas9蛋白或cpf1蛋白)。所述指导序列可以是核糖核酸指导序列。在某些方面,所述指导序列具有约10个碱基对至约150个碱基对的长度。所述一个或多个指导序列可以包括两个或更多个指导序列。
在一些实施方案中,在所述细胞中修饰2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个或20个基因组序列。所述细胞可以是干细胞、神经元、有丝分裂后细胞或成纤维细胞。在一些方面,所述细胞是人类细胞或小鼠细胞。
在一些方面,一个或多个核定位序列融合在所述无催化活性的位点特异性核酸酶与所述效应子结构域之间。在某些方面,所述基因组序列中的一个或多个与疾病或疾患有关。
在某些方面,所述方法还包括使所述细胞与抑制或增强dna甲基化的试剂接触。所述试剂可以是小分子。举例来说,所述试剂是5-氮杂胞苷或5-氮杂脱氧胞苷。
在某些实施方案中,所述方法还包括将所述细胞引入非人类哺乳动物体内。所述非人类哺乳动物可以是小鼠。
还公开了通过本文所述的方法产生的分离的修饰细胞。
本文还公开了治疗有需要的患者的方法,所述方法包括将本文所述的修饰细胞向需要这样的细胞的患者施用。
还公开了在有需要的个体中调节引起疾病的一个或多个基因组序列的甲基化的方法,所述方法包括向所述个体中引入与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶;以及一个或多个指导序列,从而在所述个体中调节引起疾病的一个或多个基因组序列的甲基化。
还公开了具有修饰的基因组的修饰细胞,所述修饰的基因组包含其中基因组序列的甲基化已经受到调节的第一基因组修饰,其中所述调节通过使细胞与和具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶和一个或多个指导序列接触而发生。
本文还公开了调节细胞中一个或多个基因组序列的甲基化的方法,所述方法包括使所述细胞与编码包含与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽的核酸;以及指导序列或编码指导序列的核酸接触。
本文还公开了在个体中调节一个或多个基因组序列的甲基化的方法,所述方法包括向所述个体施用编码包含与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽的核酸;以及指导序列或编码指导序列的核酸。
在一些方面,所述指导序列使所述多肽靶向到所述一个或多个基因组序列。所述基因组序列可以包括差异甲基化区域、增强子、启动子或ctcf结合位点。在某些方面,所述方法包括调节细胞中至少两个基因组序列的甲基化,其中所述基因组序列选自差异甲基化区域、增强子、启动子以及ctcf结合位点。
在一些实施方案中,所述效应子结构域包括tet1或dnmt3a。在一些方面,所述无催化活性的位点特异性核酸酶是无催化活性的cas蛋白(例如dcas9蛋白)。
在某些方面,所述方法还包括向所述个体施用抑制或增强dna甲基化的试剂。所述试剂可以是小分子。举例来说,所述试剂是5-氮杂胞苷或5-氮杂脱氧胞苷。
本文还公开了治疗有需要的患者的方法,所述方法包括向所述患者施用编码包含与具有甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽的核酸;以及指导序列或编码指导序列的核酸。
还公开了调节细胞中一个或多个所关注的基因的表达的方法,其中差异甲基化区域位于与所述基因的转录起始位点相距50kb以内,所述方法包括使所述细胞与编码包含与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽的核酸;指导序列或编码指导序列的核酸接触,其中所述指导序列使所述多肽靶向到所述差异甲基化区域。
在一些方面,所述差异甲基化区域在所述细胞中是过度甲基化的并且所述效应子结构域(例如tet1)具有脱甲基化活性。在其它方面,所述差异甲基化区域在所述细胞中是非甲基化的并且所述效应子结构域(例如dnmt3a)具有甲基化活性。在一些实施方案中,所述细胞是干细胞、有丝分裂后细胞、神经元或成纤维细胞。
本文还公开了鉴定甲基化状态影响所关注的基因的表达的基因组序列的方法,所述方法包括使细胞与编码包含与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽的核酸;指导序列或编码指导序列的核酸接触,其中所述指导序列使所述多肽靶向到候选基因组序列;以及测量所述基因的表达,其中如果所述与所述核酸接触的细胞中所述基因的表达不同于未与所述核酸接触的对照细胞中所述基因组区域的甲基化水平,那么将所述基因组序列鉴定为甲基化状态影响所述所关注的基因的表达的基因组序列。
在一些方面,所述基因组序列包括差异甲基化区域、增强子、启动子或ctcf结合位点。在某些方面,所述方法包括调节选自以下的至少两个基因组序列的甲基化:差异甲基化区域、增强子、启动子以及ctcf结合位点。所述一个或多个基因组序列可以位于与所述基因的转录起始位点(tss)相距50kb以内。
在某些方面,所述效应子结构域具有甲基化活性。举例来说,所述效应子结构域是dnmt3a。在其它方面,所述效应子结构域具有脱甲基化活性。举例来说,所述效应子结构域是tet1。在一些方面,所述细胞是干细胞、有丝分裂后细胞、神经元或成纤维细胞。在某些实施方案中,一个或多个核定位序列融合在所述包含所述无催化活性的位点特异性核酸酶的多肽与所述效应子结构域之间。
本文还公开了以下方法,所述方法包括根据本文所述的方法鉴定甲基化状态影响所关注的基因的表达的基因组区域;使细胞与测试剂接触;以及测量所述细胞中所述所鉴定的基因组区域的甲基化,其中如果所述与所述测试剂接触的细胞中所述基因组区域的甲基化水平不同于未与所述测试剂(例如小分子)接触的对照细胞中所述基因组区域的甲基化水平,那么将所述测试剂鉴定为所述基因组区域的甲基化调节剂。
本发明的上述和许多其它特征以及伴随的优势将通过参考以下对本发明的详细描述更好地理解。
附图说明
本专利或申请文件含有以彩色制成的至少一个附图。具有彩色附图的本专利或专利申请公布的副本将在请求和支付必要费用后由专利局提供。
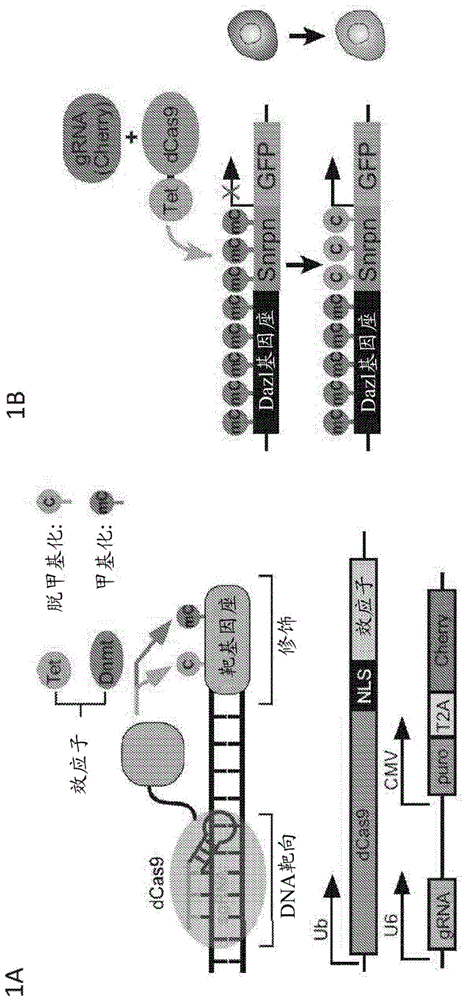
图1a-1f描绘了dcas9-tet1对dazl-snrpn-gfp报告基因的激活。图1a上图:提供了与用于消除dna甲基化的tet1融合和与用于特定序列的从头甲基化的dnmt3a融合的无催化活性的突变型cas9(dcas9)的示意图。下图:示出了具有使dcas9与tet1连接的核定位信号(nls)的优化的dcas9-效应子构建体和具有puro盒和cherry盒的指导rna构建体。图1b是使用特异性grna使dcas9-tet1靶向snrpn启动子区域以消除甲基化并且激活gfp表达的示意图。图1c示出了将dazl-snrpn-gfpmesc用表达dcas9-tet1(dc-t)和乱序grna(scgrna)或靶向snrpn启动子区域的4个grna(靶grna)的慢病毒感染。通过在感染后3天对这些细胞进行流式细胞术分析来计算gfp阳性细胞的百分比,并且显示为两次生物平行测定的平均gfp阳性细胞百分比±sd。请注意,gfp阳性细胞的百分比被表示为占受感染的cherry阳性细胞的分数。图1d左侧:示出了在培养1周之后图1c中的分选的cherry阳性细胞的代表性荧光图像。比例尺:250um。右侧:提供了对gfp阳性集落的百分比进行定量,并且显示为两次生物平行测定的平均gfp阳性细胞百分比±sd。图1e示出了图1c中所述的细胞的亚硫酸氢盐测序。图1f提供了snrpn启动子区域和相邻dazl基因座中单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd。
图2a-2h描绘了dcas9-dnmt3a使gapdh-snrpn-gfp报告基因沉默。图2a示出了使用特异性grna使dcas9-dnmt3a靶向snrpn启动子区域以将启动子甲基化并且使gfp表达沉默的示意图。图2b示出了将gapdh-snrpn-gfpmesc用表达dcas9-dnmt3a(dc-d)和乱序grna(scgrna)或靶向snrpn启动子区域的grna(靶grna)的慢病毒感染。通过在感染后3天进行流式细胞术分析来计算gfp阴性细胞的百分比,并且显示为两次生物平行测定的平均gfp阴性细胞百分比±sd。请注意,gfp阳性细胞的百分比被表示为占受感染的cherry阳性细胞的分数。图2c左侧:示出了在培养1周之后b中的分选的cherry阳性细胞的代表性荧光图像。比例尺:250um。右侧:示出了对gfp阴性集落的百分比进行定量,并且显示为两次生物平行测定的平均gfp阴性细胞百分比±sd。图2d提供了图2b中所述的细胞的亚硫酸氢盐测序。图2e描绘了snrpn启动子区域和相邻gapdh基因座中单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd。图2f示出了将具有多西环素(doxycycline)诱导型dcas9-dnmt3a的gapdh-snrpn-gfpmesc在多西环素(2ug/ml)存在下用表达靶向snrpn启动子区域的grna的慢病毒感染。通过在感染后3天进行流式细胞术分析来计算gfp阴性细胞的百分比,并且显示为两次生物平行测定的平均gfp阴性细胞百分比±sd。请注意,gfp阳性细胞的百分比被表示为占受感染的cherry阳性细胞的分数。图2g左侧:描绘了在存在或不存在多西环素的情况下培养1周之后图2f中的分选的cherry阳性群体的代表性荧光图像。比例尺:250um。右侧:示出了对gfp阴性集落的百分比进行定量,并且显示为两次生物平行测定的平均gfp阴性细胞百分比±sd。图2h描绘了来自图2g中的细胞的snrpn启动子区域和相邻gapdh基因座中每一个单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd。
图3a-3e描绘了dcas9-tet1对bdnf启动子iv的靶向脱甲基化以激活神经元中的bdnf。图3a提供了使用特异性grna使dcas9-tet1(dc-t)靶向bdnf启动子iv以消除甲基化并且激活bdnf表达的示意图。图3b示出了将在体外培养3天(div3)的小鼠皮层神经元用在存在或不存在靶向bdnf启动子iv的grna的情况下表达dc-t或表达tet1的无催化活性形式(dc-dt,在h1672y和d1674a处具有突变)和bdnfgrna的慢病毒感染2天,然后使用或不使用kcl(50mm)处理6小时,之后收获用于rt-qpcr分析。条柱是三次生物平行测定的平均值±sd。图3c提供了图3b中的bdnf诱导的代表性共聚焦图像。上图:通过50mmkcl处理诱导bdnf表达6小时。下图:当dc-t与靶向bdnf启动子iv区域的grna共表达时显著诱导bdnf表达。请注意,dc-dt与bdnfgrna的共表达没有激活bdnf表达。针对map2(两个上图)或cherry(两个下图)以红色染色,针对bdnf以绿色染色,针对dapi以蓝色染色并且针对dcas9以灰色染色。比例尺:50um。图3d描绘了图3c中的神经元的亚硫酸氢盐测序。图3e示出了bdnf启动子iv区域中每一个单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd。
图4a-4h描绘了dcas9-tet1对myod远端增强子进行靶向脱甲基化以促进成纤维细胞转化成成肌细胞。图4a提供了使用特异性grna使dcas9-tet1(dc-t)靶向dmr-5中的myod远端增强子(de)区域的示意图。图4b示出了将c3h10t1/2小鼠胚胎成纤维细胞用表达dc-t和靶grna或tet1的无催化活性形式(dc-dt)和靶grna的慢病毒感染2天。对cherry阳性细胞进行facs分选用于rt-qpcr分析。条柱表示三次实验平行测定的平均值±sd。图4c是图4b中的细胞的亚硫酸氢盐测序。图4d示出了myodde区域中单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd。图4e示出了在成纤维细胞向成肌细胞转化测定中在第14天时c3h10t1/2细胞的代表性共聚焦图像。将c3h10t1/2细胞以每孔1×104个细胞平板接种到6孔板中,然后用表达dc-t和靶向dmr-5的grna或tet1的无催化活性形式(dc-dt)和靶grna的慢病毒感染。在感染后24小时,将细胞用媒介物对照(hepes缓冲液)或5-氮杂胞苷(1um)处理24小时,然后在14天之后收获用于免疫荧光染色。针对myod以绿色染色,针对mhc以洋红色染色并且针对dapi以蓝色染色。比例尺:200um。图4f提供了在用表达单独的dc-t、dc-t和靶向dmr-5的grna以及dc-dt和靶grna的慢病毒感染后14天时myod阳性细胞比率的定量。图4g示出了在感染后14天时基于每个mhc+簇的核数(被分组为每个mhc+簇2-5个、6-10个、11-20个以及>20个核)mhc阳性细胞簇的分布图。当用5-aza处理时,与模拟对照或单独的dc-t相比,dc-t和靶grna的共表达显著促进更加成熟的更大的mhc+簇的形成,但是其它组合不是这样。图4h提供了在感染后14天时具有超过2个或5个核的mhc阳性簇中肌管密度的定量。添加5-aza诱导了mhc+肌管形成。dc-t和靶grna的共表达与5-aza显著协同作用,从而诱导更多的和更大的肌管(>5个核的mhc+簇)。对于f-h,数据是从3-5个代表性图像中定量的。条柱表示平均值±sd。
图5a-5i描绘了ctcf结合位点的靶向甲基化。图5a提供了使用特异性grna使dcas9-dnmt3a靶向ctcf结合位点以诱导从头甲基化,阻断ctcf募集,并且打开ctcf环,从而改变相邻环中的基因表达的示意图。图5b提供了在环中具有超级增强子和mir290的ctcf靶-1(mir290基因座)、左侧相邻环中的au018091基因以及右侧相邻环(靠近所靶向的ctcf结合位点)中的nlrp12基因的示意图。myadm基因在含有nlrp12的环的右侧的相邻环中。超级增强子结构域被示为红色条。所靶向的ctcf位点用方框突出显示。还示出了ctcf(黑色)和h3k27ac(超级增强子)(红色)的chip-seq结合谱(每百万个读段中每个碱基对的读段数)以及甲基化轨迹(黄色),其中dmr呈蓝色。图5c-5e示出了将多西环素诱导型dcas9-dnmt3amesc用表达乱序grna或ctcf靶-1grna以及dc-dt和ctcf靶-1grna的慢病毒感染3天,然后对cherry阳性细胞进行facs分选。在多西环素存在下培养3天之后,将这些细胞平板接种到明胶包被的板上1小时以去除饲养细胞,然后收获用于进行图5c中的rt-qpcr分析、图5d和图5e中的亚硫酸氢盐测序分析。条柱表示三次实验平行测定的平均值±sd。图5f提供了在该环中具有超级增强子和pou5f1基因的ctcf靶-2、左侧相邻环(靠近所靶向的ctcf结合位点)中的h2q10基因以及右侧相邻环中的tcf19的示意图。超级增强子结构域被示为红色条。所靶向的ctcf位点用方框突出显示。还示出了ctcf(黑色)和h3k27ac(超级增强子)(红色)的chip-seq结合谱(每百万个读段中每个碱基对的读段数)以及甲基化轨迹(黄色),其中dmr呈蓝色。图5g-5i示出了如图5c-5e中所述针对ctcf靶-2进行的同一组实验,并且收获细胞用于进行如图5c中的rt-qpcr分析和如图5d和图5e中的亚硫酸氢盐测序。条柱表示三次实验平行测定的平均值±sd。
图6a-6d描绘了ctcf结合位点的靶向甲基化以操纵ctcf环。图6a提供了在mir290基因座处对c中所述的细胞进行的定量染色体构象捕获(3c)分析。超级增强子结构域被示为红色条。所靶向的ctcf位点用方框突出显示。箭头指示染色体位置,在所述染色体位置之间测定了相互作用频率。星号指示3c锚定位点。还示出了ctcf(黑色)和h3k27ac(超级增强子)(红色)的chip-seq结合谱(每百万个读段中每个碱基对的读段数)以及甲基化轨迹(黄色),其中dmr呈蓝色。所示的染色体位置与3c锚定位点之间的相互作用频率被示为下图上的条形图(平均值±sd)。qpcr反应按一式两份运行,并且值相对于具有乱序grna的细胞中的平均相互作用频率标准化。(对于所有三个区域,p<0.05;司图顿氏t检验(student'sttest),ns表示无显著性,nc表示阴性对照。)图6b示出了使用图6a中的细胞进行抗ctcfchip实验,之后进行定量pcr分析。条柱表示三次实验平行测定的平均值±sd。图6c提供了在pou5f1基因座处对图5g中所述的细胞进行的定量染色体构象捕获(3c)分析。超级增强子结构域被示为红色条。所靶向的ctcf位点用方框突出显示。箭头指示染色体位置,在所述染色体位置之间测定了相互作用频率。星号指示3c锚定位点。还示出了ctcf(黑色)和h3k27ac(超级增强子)(红色)的chip-seq结合谱(每百万个读段中每个碱基对的读段数)以及甲基化轨迹(黄色),其中dmr呈蓝色。所示的染色体位置与3c锚定位点之间的相互作用频率被示为下图上的条形图(平均值±sd)。qpcr反应按一式两份运行,并且值相对于具有乱序grna的细胞中的平均相互作用频率标准化。(p<0.05;司图顿氏t检验,ns表示无显著性。)。图6d示出了使用c中的细胞进行抗ctcfchip实验,之后进行定量pcr分析。条柱表示三次实验平行测定的平均值±sd。
图7a-7h描绘了由dcas9-tet1进行靶向离体和体内dna甲基化编辑以激活沉默的gfp报告基因。图7a提供了图示了离体激活小鼠成纤维细胞中沉默的gfp报告基因的实验程序的示意图。小鼠尾部成纤维细胞源自于在dlk1-dio3基因座中携带父本ig-dmr-snrpn-gfp等位基因(ig-dmrgfp/pat)的遗传修饰的小鼠品系。父本等位基因上的ig-dmr-snrpn启动子是过度甲基化的,因此gfp报告基因是组成型沉默的。将培养的成纤维细胞用表达dcas9-tet1和grna的慢病毒载体感染以将snrpn启动子脱甲基化并且激活gfp报告基因。对细胞进行成像和facs分析。图7b示出了用表达dcas9-tet1(dc-t)和scgrna、dcas9-tet1的无活性形式(dc-dt)和snrpn靶grna或dcas9-tet1和snrpn靶grna的慢病毒感染的ig-dmrgfp/pat成纤维细胞的代表性免疫组织化学图像。针对cherry以红色染色,针对gfp以绿色染色并且针对核以dapi染色。比例尺:100um。请注意,为了开启snrpn-gfp甲基化报告基因,必须将dcas9-tet1和靶grna慢病毒载体都转导到相同的细胞中。因此,预期在该实验中cherry阳性细胞(靶grna)的数量大于gfp阳性细胞(snrpn启动子的脱甲基化)的数量。图7c提供了在cherry(grna)阳性细胞中具有gfp激活的ig-dmrgfp/pat小鼠成纤维细胞的百分比的定量。约80%的具有snrpn靶grna表达的细胞开启gfp表达。dc-t和scgrna以及dc-t的无活性形式(dc-dt)和snrpn靶grna不能开启gfp报告基因表达。条柱表示三次实验平行测定的平均值±sd。图7d提供了图示了体内激活ig-dmrgfp/pat小鼠脑中的gfp报告基因的实验程序的示意图。使用立体定位显微注射方法递送表达dc-t和scgrna、dc-dt和snrpn靶grna以及dc-t和snrpn靶grna的慢病毒载体。将脑切片并且通过免疫组织化学方法分析。图7e提供了用dc-t和scgrna、dc-dt和snrpn靶grna以及dc-t和snrpn靶grna感染的ig-dmrgfp/pat小鼠脑的代表性共聚焦显微照片。只有dc-t和靶grna激活了gfp表达。比例尺:100um。图7g-7h提供了在脑(图7g)和皮肤表皮(图7h)中体内慢病毒递送实验中cherry(grna)阳性细胞中具有gfp激活的ig-dmrgfp/pat细胞的百分比的定量。被转导有snrpn靶grna表达的约70%的神经元和85%的皮肤真皮细胞开启体内gfp表达。相反,dc-t和乱序grna以及tet1的无活性形式(dc-dt)和snrpn靶grna没有激活gfp报告基因表达。条柱表示来自2只动物的超过四个代表性图像的平均值±sd。
图8a-8e描绘了用于编辑哺乳动物基因组中的5-胞嘧啶dna甲基化的修饰的crispr系统。图8a描绘了具有位于不同位置处的核定位信号(nls)的dcas9-效应子构建体以及具有cmv驱动的puro-t2a-cherry盒的指导rna(grna)或增强型指导rna(e-grna)的设计。ub:人类泛素c启动子。图8b示出了在hek293t细胞中用这些构建体转染2天之后通过用抗cas9抗体进行免疫印迹来分析dcas-nls-tet1和nls-dcas9-nls-tet1的表达。使用α-微管蛋白作为上样对照。图8c提供了在存在或不存在靶向myod基因座中的相同位置的grna或e-gnra的共表达的情况下,hek293t细胞中dcas9-nls-tet1和nls-dcas9-nls-tet1的细胞定位的比较。在不存在sgrna的情况下,dcas9-nls-tet1主要被排除在核隔室之外,并且nls-dcas9-nls-tet1在转染的hek293t细胞中显示出弱的核定位。grna或e-grna的共表达诱导了这两种蛋白质的细胞质向核的易位。在合并图像中针对dcas9以绿色染色,针对cherry以红色染色并且针对dapi以蓝色染色。前两个图中的红色虚线指示用于gfp强度定量的图像的横截面。比例尺:10um。图8d在盒须图中提供了在不存在或存在grna或e-gnra的情况下hek293t细胞中dcas9-nls-tet1和nls-dcas9-nls-tet1的核-细胞质比的定量。使用沿着如c中所示的横截面线的细胞质和核结构域的平均dcas9强度进行定量。“+”表示每一组中20个数据点的平均值;盒指示极端数据点(上棒和下棒)、25%-75%区间(盒)以及中值(中心线)。图8e示出了dcas9-nls-tet1和nls-dcas9-nls-tet1的诱导指数(被定义为相对于在不存在sgrna的情况下的核-细胞质比标准化的在sgrna存在下的核-细胞质比)的定量。grna和e-grna分别将dcas9-nls-tet1的核定位诱导到3.17倍和3.22倍并且将nls-dcas9-nls-tet1的核定位诱导到1.73倍和1.77倍。我们推论具有最高诱导指数的组合将产生最佳的靶向dna甲基化编辑的信噪比。
图9a-9j描绘了进行靶向启动子甲基化编辑以由dcas9-tet1激活dazle-snrpn-gfp报告基因并且由dcas9-dnmt3a阻遏gapdh-snrpn-gfp报告基因。图9a提供了dazl-snrpn基因座的基因组序列,其中grna序列以黄色标记并且cpg以绿色标记。每一个grna的pam由红色方框突出显示。dazl序列以小写字母表示,并且snrpn序列以大写字母表示。图9b提供了用在存在或不存在针对snrpn启动子的靶grna的情况下表达dcas9-tet1(dc-t)的慢病毒感染3天的dazl-snrpn-gfpmesc的荧光图像。比例尺:120um。图9c提供了在用慢病毒感染以表达dcas9-tet1(dc-t)和乱序grna或靶向snrpn启动子区域的4个grna后3天时dazl-snrpn-gfpmesc的流式细胞术分析。通过所列的方程式计算激活效率并且显示为两次生物平行测定的平均cherry和gfp双阳性细胞百分比±sd。图9d描绘了在对用表达dc-t和snrpngrna的慢病毒感染的dazl-snrpn小鼠es细胞进行facs分选之后cherry+;gfp+细胞群体或cherry+;gfp-细胞群体中的dazl-snrpn区域的亚硫酸氢盐测序。图9e提供了gapdh-snrpn基因座的基因组序列,其中grna序列以黄色标记并且cpg以绿色标记。每一个grna的pam由红色方框突出显示。gapdh序列以小写字母表示,并且snrpn序列以大写字母表示。图9f提供了在用慢病毒感染以表达dcas9-dnmt3a(dc-d)和靶向snrpn启动子区域的3个grna后3天时gapdh-snrpn-gfpmesc的流式细胞术分析。通过所列的方程式计算失活效率并且显示为两次生物平行测定的平均cherry阳性和gfp阴性细胞百分比±sd。图9g描绘了在对用表达dc-d和snrpngrna的慢病毒感染的gapdh-snrpn小鼠es细胞进行facs分选之后cherry+;gfp+细胞群体或cherry+;gfp-细胞群体中的gapdh-snrpn区域的亚硫酸氢盐测序。图9h示出了在多西环素(2ug/ml)处理48小时之后通过rt-qpcr分析具有稳定整合的多西环素诱导型dcas9-dnmt3a盒的mesc。条柱表示三次实验平行测定的平均值±sd。图9i提供了在多西环素(2ug/ml)存在下用表达与f中的相同的3个grna的慢病毒感染3天后具有多西环素诱导型dcas9-dnmt3a的gapdh-snrpn-gfpmesc的流式细胞术分析。如底部处所示计算失活效率并且表示为两次生物平行测定的平均cherry阳性和gfp阴性细胞百分比±sd。图9j左图:示出了dcas9-dnmt3a-p2a-bfp构建体和grna-cherry构建体的示意图。中图:提供了在用表达dcas9-dnmt3a-p2a-bfp和snrpngrna的慢病毒感染之后通过对gapdh-snrpn-gfpmesc进行facs分析所获得的仅bfp阳性细胞群体、仅cherry阳性细胞群体以及双阳性细胞群体的百分比。右图:描绘了bfp+;cherry+细胞群体内gfp-细胞或gfp+细胞的百分比的facs分析。
图10a-10g描绘了基于tale的甲基化编辑和基于dcas9的甲基化编辑的比较。图10a示出了将hela细胞用dcas9-dnmt3a和一个p16靶grna(cherry)或tale-dnmt3a-gfp转染。在转染后48小时对转染阳性细胞群体(cherry+)或(gfp+)进行facs分选。通过亚硫酸氢盐测序来分析p16启动子区域中每一个单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd,其中针对dcas9-dnmt3a对总共34个单一集落进行测序,并且针对tale-dnmt3a对总共31个单一集落进行测序。红色箭头指示p16靶grna的位置,并且紫色箭头指示tale-dnmt3a的结合位点。图10b描绘了将hek293t细胞用dcas9-tet1和一个rhoxf2靶grna(具有puro盒)或具有puro盒表达质粒的tale-tet1共转染。将嘌呤霉素(2ug/ml)添加到培养基中以选择转染阳性细胞。在选择2天之后收获细胞用于通过亚硫酸氢盐测序分析rhoxf2启动子区域中单个cpg的甲基化水平。示出了两次生物平行测定的平均百分比±sd,其中针对dcas9-tet1对总共34个单一集落进行测序并且针对tale-tet1对总共38个单一集落进行测序。红色箭头指示rhoxf2靶grna的位置,并且紫色箭头指示tale-dnmt3a的结合位点。图10c提供了a和b中的甲基化水平分析的汇总。通过由dcas9-dnmt3a/tet1显著编辑(甲基化变化大于10%)的cpg与grna靶向位点相距的距离来确定有效范围。分辨率被定义为dcas9-dnmt3a/tet1和一个单一grna的有效范围,并且更好的分辨率指的是dcas9-dnmt3a/tet1的更短有效范围,这将允许更精确地编辑dna甲基化。图10d示出了将图9h中所述的dox诱导型dcas9-dnmt3a表达小鼠es细胞用乱序grna或靶向mir290基因座或dazl-snrpn基因座的grna感染。将facs分选的cherry阳性细胞与dox(2ug/ml)一起培养3天。然后,收获这些细胞用于进行抗dcas9chip-seq分析。使用先前所述的成对峰呼叫程序来呼叫峰(wu等,2014),并且呈现于描绘全基因组chip-seq峰的曼哈顿图(manhattanplot)中。显示出所有具有p<0.001的峰。每一个点表示峰,其中x轴显示基因组位置并且y轴显示由基于模型的chip-seq分析(macs)输出的峰顶高度(zhang等,2008)。每一个点的大小与它的y轴值成比例,并且单个染色体的颜色不同以用于可视化。图10e描绘了说明具有最高信号水平的靶向基因座(mir290或dazl-snrpn)和具有第二高信号和第三高信号的两个脱靶基因座(对于mir290grna,是vac14基因座和tenm4基因座;对于dazl-snrpngrna,是vrk1基因座和gm42619基因座)处的chip-seq峰,其中邻近基因列于下方。请注意,4个dazl-snrpngrna识别如图9a中所述的dazl和snrpn的启动子序列,因此这组grna的峰被定位到这两个基因座。图10f示出了对来自图5c中所用的细胞的基因组dna进行vac14基因座和tenm4基因座处脱靶结合位点的亚硫酸氢盐测序。示出了两次生物平行测定的平均百分比±sd。图10g示出了对来自图1d和图2c中所用的细胞的基因组dna进行vrk1基因座和gm42619基因座处脱靶结合位点的亚硫酸氢盐测序。示出了两次生物平行测定的平均百分比±sd。
图11a-11l描绘了在神经元中dcas9-tet1对bdnf启动子iv进行靶向脱甲基化。图11a示出了bdnf启动子iv区域的基因组序列,其中grna序列以黄色标记并且cpg以绿色标记。每一个grna的pam由红色方框突出显示。图11b左图:示出了描绘了对e17.5小鼠原代皮层神经元进行kcl处理和慢病毒递送实验以研究bdnf表达的示意图。请注意,在div2时用arac处理培养的神经元以停止神经胶质细胞和神经祖细胞中的细胞分裂。右图:示出了用50mmkcl处理div3小鼠皮层神经元,并且在不同的时间点收获用于通过rt-qpcr进行bdnf表达分析。图11c提供了在kcl处理24小时的过程中小鼠原代神经元的edu标记分析。请注意,观测到非常少的edu阳性细胞。针对edu以红色染色,针对map2以绿色染色并且针对核以dapi染色。比例尺:500um。图11d上图:提供了在kcl处理过程中神经元密度的定量。有丝分裂后神经元的密度随时间推移稳定保持在约4.5×104个/cm2。条柱表示三次实验平行测定的平均值±sd。下图:提供了在kcl处理的过程中edu阳性细胞的定量。在24小时内少于2%的细胞呈edu阳性。条柱表示三次实验平行测定的平均值±sd。图11e提供了由dcas9-tet1和一组4个靶向bdnf启动子iv的grna的异位表达实现的bdnf诱导的共聚焦显微照片。针对bdnf以绿色染色,针对map2以洋红色染色,针对cherry以红色染色并且针对dapi以蓝色染色。请注意,在这些神经元中慢病毒感染效率接近100%。比例尺:50um。图11f示出了对从b和e中收获的神经元进行npas4表达的rt-qpcr分析。条柱表示三次实验平行测定的平均值±sd。图11g示出了将div3小鼠皮层神经元用单独的dc-t或dc-t和靶向bdnf启动子iv的4个grna或单个bdnfgrna感染,然后进行bdnf表达的qpcr分析。图11h-11i示出了用表达dc-t或dc-dt和4个bdnfgrna的慢病毒感染40小时和60小时的神经元(h)或在kcl处理6小时之后的神经元(图11i)的tet辅助亚硫酸氢盐测序(tab-seq)分析。图11j描绘了将div3小鼠皮层神经元用abt(50um)处理6小时,然后用kcl(50mm)处理6小时,之后收获用于进行rt-qpcr分析。条柱表示三次实验平行测定的平均值±sd。图11k描绘了将div3小鼠皮层神经元用2-羟基戊二酸盐(10mm)处理2小时,然后用kcl(50mm)处理6小时,之后收获用于进行rt-qpcr分析。条柱表示三次实验平行测定的平均值±sd。图11l描绘了对源自于野生型或tet1敲除e17.5胚胎的div3小鼠皮层神经元进行时间过程kcl处理实验(6小时、9小时以及12小时)。条柱表示三次实验平行测定的平均值±sd。
图12a-12j描绘了dcas9-tet1对myoddmr-5进行的靶向脱甲基化以及成纤维细胞向成肌细胞的转化。图12a示出了myod远端增强子区域的基因组序列,其中grna序列以黄色标记并且cpg以绿色标记。每一个sgrna的pam由红色方框突出显示。图12b提供了成纤维细胞向成肌细胞转化测定的实验方案。简单地说,将c3h10t1/2小鼠胚胎成纤维细胞以每孔1×104个细胞平板接种到6孔板中,然后用表达dcas9-tet1和靶grna的慢病毒感染。在感染后24小时,任选地将细胞用5-氮杂胞苷(1um)处理24小时(以红色标记),并且在不同的时间点(第14天、第16天以及第25天,以深蓝色标记)收获用于进行免疫荧光染色,其中每隔一天更换培养基。比例尺:100um。图12c示出了在5-aza处理之后c3h10t1/2成纤维细胞的肌管形成的代表性共聚焦显微照片。上图:示出了克隆视野含有稀疏分布的小尺寸和中尺寸肌管。中图:示出了克隆视野含有稀疏分布的大尺寸肌管。下图:示出了克隆视野含有高密度的具有异质尺寸的肌管。针对mhc以绿色染色,针对myod以红色染色并且针对dapi以蓝色染色。比例尺:200um。图12d描绘了在5-aza处理之后不同的时间表达myod的模拟c3h10t1/2细胞的分数。表达myod的细胞的分数从第14天时的约6%增加到第16天时的约13%,并且在第25天时达到约20%。条柱表示平均值±sd。图12e提供了mhc+细胞簇中的核数(被分组为每个mhc+簇1-2个和>2个核)。在5-aza处理之后的随后时间点观测到更大肌管的形成。条柱表示平均值±sd。在图12d和图12e中数据是从每一组的3-5个代表性图像中定量的。图12f示出了将c3h10t1/2细胞用表达dc-t和myodgrna的慢病毒感染24小时,并且使用或不使用5-aza处理48小时,之后收获用于进行qpcr分析。条柱表示三次实验平行测定的平均值±sd。图12g提供了在如图12b中所述的成纤维细胞向成肌细胞转化测定中用表达dc-t和靶向dmr-5(myod远端增强子)的grna的慢病毒感染之后16天时c3h10t1/2细胞的代表性图像。请注意,在具有dc-t和靶grna的细胞中观测到适度水平的myod激活(与用5-aza处理的细胞相比),但是没有观测到肌球蛋白重链(mhc)表达或肌管形成。针对mhc以洋红色染色,针对myod以绿色染色并且针对dapi以蓝色染色。比例尺:200um。图12h描绘了在用表达单独的dc-t或dc-t和靶向dmr-5的grna的慢病毒感染后16天时myod阳性细胞的分数。图12i示出了在感染后16天时mhc+细胞簇中的核数(被分组为每个mhc+簇2-5个、6-10个、11-20个以及>20个核)。当用5-aza处理时,与模拟对照或单独的dc-t相比,grna和dc-t的共表达显著促进更大的和更加成熟的mhc+簇的形成。图12j示出了在感染后16天时具有超过2个或5个核的mhc阳性簇的肌管密度。添加5-aza诱导了mhc+肌管形成。dc-t和grna的共表达显著诱导更多的和更大的肌管(>5个核的mhc+簇)。对于图12h-12j,数据是从3-5个代表性图像中定量的。条柱表示平均值±sd。
图13a-13b描绘了用于mir290基因座和pou5f1基因座的ctcf结合位点的靶向甲基化的grna设计。图13a提供了ctcf靶1区域(mir290基因座)的基因组序列,其中grna序列以黄色标记并且cpg以绿色标记。每一个sgrna的pam由红色方框突出显示,预测的ctcf结合基序在蓝色方框中突出显示。图13b提供了ctcf靶2区域(h2q10-pou5f1基因座)的基因组序列,其中grna序列以黄色标记并且cpg以绿色标记。每一个sgrna的pam由红色方框突出显示,预测的ctcf结合基序在蓝色方框中突出显示。
图14a-14f描绘了在小鼠皮肤细胞中由dcas9-tet1介导的脱甲基化激活ig-dmrgfp/pat报告基因。图14a提供了图示了离体激活ig-dmrgfp/pat小鼠成纤维细胞中沉默的gfp报告基因的实验程序的示意图。将培养的成纤维细胞用表达dcas9-tet1和grna的慢病毒载体感染以将snrpn启动子脱甲基化并且激活gfp报告基因。图14b提供了受感染的ig-dmrgfp/pat小鼠成纤维细胞的facs分析。图14c示出了图14b中的cherry阳性细胞群体中gfp+细胞百分比的定量。图14d提供了图示了对ig-dmrgfp/pat小鼠的腹侧上的每一个部位的慢病毒递送方法的示意图。图14e提供了用dcas9-tet1和scgrna、tet1的无活性形式(dc-dt)和snrpngrna以及dcas9-tet1和snrpngrna感染的ig-dmrgfp/pat小鼠皮肤的代表性共聚焦显微照片。箭头指示只有dc-t和snrpngrna激活gfp表达。请注意表皮左边缘的红色自发荧光。图14f提供了在慢病毒递送dc-t和snrpngrna以激活gfp表达的情况下2个毛囊的代表性共聚焦显微照片。
图15示出了细胞类型的各种实例。
图16描绘了dna甲基化。
图17示出了包括dna甲基化和逆转dna甲基化的dna甲基化循环。
图18提供了人类组织的dna甲基化图谱的实例。
图19a-19e描绘了甲基化活性和脱甲基化活性的汇总。图19a示出了dazi-snrpn-gfp报告基因的脱甲基化和gapdh-snrpn-gfp报告基因的甲基化。图19b示出了dcas9-tet1对bdnf启动子iv进行靶向脱甲基化以激活bdnf。图19c示出了dcas9-tet1对myod远端增强子进行靶向脱甲基化以促进肌细胞转分化。图19d示出了ctcf结合位点的靶向甲基化。图19e示出了由dcas9-tet1进行靶向体内dna甲基化编辑以激活沉默的gfp报告基因。
图20描绘了在脆性x综合征中fmr-1的过度甲基化的逆转。使表现出脆性x综合征的细胞与dcas9-tet1融合蛋白接触以特异性地将ccg过度甲基化进行脱甲基化以重新激活fmr-1。
图21示出了在有丝分裂后神经元中操作bdnf脱甲基化的提议。
图22描绘了在用表达单独的dc-t、dc-t和靶grna以及dc-dt和靶grna的慢病毒感染之后myod阳性细胞比率的定量。
图23a-23c描绘了ctcf结合位点的靶向甲基化。图23a示出了染色体的三维结构。图23b示出了通过qrt-pcr测量的野生型细胞和ctcf位点缺失型细胞中所示基因的基因表达水平。图23c示出了使用c中的细胞进行抗ctcfchip实验,之后进行定量pcr分析。条柱表示三次实验平行测定的平均值±sd。
具体实施方式
除非另外指明,否则本发明的实施通常将使用细胞生物学、细胞培养、分子生物学、转基因生物学、微生物学、重组核酸(例如dna)技术、免疫学以及rna干扰(rnai)的常规技术,它们在本领域的技术范围内。这些技术中的某些技术的非限制性描述见于以下出版物中:ausubel,f.等(编著),currentprotocolsinmolecularbiology;currentprotocolsinimmunology;currentprotocolsinproteinscience;以及currentprotocolsincellbiology,全部都由johnwiley&sons,n.y.出版,截至2008年12月的版本;sambrook、russell以及sambrook,molecularcloning:alaboratorymanual,第3版,coldspringharborlaboratorypress,coldspringharbor,2001;harlow,e.和lane,d.,antibodies-alaboratorymanual,coldspringharborlaboratorypress,coldspringharbor,1988;freshney,r.i.,“cultureofanimalcells,amanualofbasictechnique”,第5版,johnwiley&sons,hoboken,nj,2005。关于治疗剂和人类疾病的非限制性信息见于goodmanandgilman'sthepharmacologicalbasisoftherapeutics,第11版,mcgrawhill,2005;katzung,b.(编著)basicandclinicalpharmacology,mcgraw-hill/appleton&lange;第10版(2006年)或第11版(2009年7月)中。关于基因和遗传性病症的非限制性信息见于mckusick,v.a.:mendelianinheritanceinman.acatalogofhumangenesandgeneticdisorders.巴尔的摩(baltimore):约翰·霍普金斯大学出版社(johnshopkinsuniversitypress),1998(第12版)或最新的在线数据库:onlinemendelianinheritanceinman,omimtm.约翰·霍普金斯大学(马里兰州的巴尔的摩(baltimore,md))的麦库斯克-内萨斯遗传医学研究所(mckusick-nathansinstituteofgeneticmedicine)和国家医学图书馆(nationallibraryofmedicine)(马里兰州的贝塞斯达(bethesda,md))的国家生物技术信息中心(nationalcenterforbiotechnologyinformation),截至2010年5月1日,ncbi.nlm.nih.gov/omim/;以及onlinemendelianinheritanceinanimals(omia),其是动物物种(除人类和小鼠以外)中的基因、遗传性病症以及性状的数据库,在omia.angis.org.au/contact.shtml上。本文提到的所有专利、专利申请以及其它出版物(例如科学论文、书籍、网站以及数据库)以引用的方式整体并入本文。在本说明书与所并入的参考文献中的任一篇之间有矛盾的情况下,应当以本说明书(包括其任何修改,所述修改可能基于所并入的参考文献)为准。除非另有说明,否则在本文使用术语的标准的本领域公认的含义。在本文使用各种术语的标准缩写。
在一个方面,本发明涉及一种修饰或调节细胞中的一个或多个基因组序列的方法,所述方法包括向所述细胞中引入与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶以及一个或多个指导序列。所述方法可以引起所述细胞中的一个或多个基因组序列的修饰。可以通过所述方法产生分离的修饰的细胞。所述无催化活性的位点特异性核酸酶可以与所述一个或多个指导序列中的每一个结合并且所述效应子结构域调节所述基因组序列的甲基化或脱甲基化(例如dna甲基化或dna脱甲基化)。可以将一个或多个指导序列、无催化活性的位点特异性核酸酶以及效应子结构域引入细胞、合子、胚胎或非人类哺乳动物中。
在其它方面,本发明涉及一种调节细胞中一个或多个基因组序列的甲基化的方法。所述方法可以包括使所述细胞与编码包含与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽的核酸接触。进一步使所述细胞与指导序列或编码指导序列的核酸接触。在一些方面,所述指导序列使所述多肽靶向到所述一个或多个基因组序列。在一些实施方案中,所述接触所述细胞可以包括直接引入到所述细胞中。在其它方面,所述接触所述细胞包括在所述细胞中表达或诱导所述细胞中的表达。基因组甲基化的报告基因描述于美国申请号15/078,851中,该美国申请以引用的方式整体并入本文。
有各种方式可以向细胞或受试者递送包含与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶的多肽,例如通过施用编码所述多肽的核酸,所述核酸可以是例如病毒载体或可以是可翻译的核酸(例如合成修饰的mrna)。修饰的mrna的实例描述于warren等(cellstemcell7(5):618-30,2010);mandalpk,rossidj.natprotoc.20138(3):568-82;美国专利公布号20120046346和/或pct/us2011/032679(wo/2011/130624)中。另外的实例见于modernatherapeutics公司的许多pct和美国申请和授权专利中,例如pct/us2011/046861;pct/us2011/054636;pct/us2011/054617;ussn14/390,100(以及这些中提到的另外的专利和专利申请)。此外,所述指导序列可以作为编码指导序列的核酸递送。举例来说,施用可以通过直接向组织或器官(例如皮肤、心脏、肝脏、肺、肾脏、脑、眼睛、肌肉、骨、神经)或肿瘤施用来进行。施用可以通过任何途径(例如口服、静脉内、腹膜内、管饲、局部、透皮、肌内、肠内、皮下)进行,可以是全身的或局部的,可以包括任何剂量(例如约0.01mg/kg至约500mg/kg),可以包括单次剂量或多次剂量。所述核酸可以被封装在例如脂质体、聚合物颗粒(例如plga颗粒)中。
可以使用本文所述的方法来修饰或调节多种细胞中的一个或多个基因组序列,所述细胞包括体细胞、干细胞、有丝分裂细胞或有丝分裂后细胞、神经元、成纤维细胞或合子。细胞、合子、胚胎或出生后哺乳动物可以是脊椎动物(例如哺乳动物)来源的。在一些方面,所述脊椎动物是哺乳动物或禽类。具体实例包括灵长类动物(例如人类)、啮齿类动物(例如小鼠、大鼠)、犬、猫、牛、马、羊、猪或禽类(例如鸡、鸭、鹅、火鸡)细胞、合子、胚胎或出生后哺乳动物。在一些实施方案中,所述细胞、合子、胚胎或出生后哺乳动物是分离的(例如分离的细胞;分离的合子;分离的胚胎)。在一些实施方案中,使用小鼠细胞、小鼠合子、小鼠胚胎或小鼠出生后哺乳动物。在一些实施方案中,使用大鼠细胞、大鼠合子、大鼠胚胎或大鼠出生后哺乳动物。在一些实施方案中,使用人类细胞、人类合子或人类胚胎。可以使用本文所述的方法在哺乳动物(例如小鼠)体内修饰或调节一个或多个基因组序列(例如将基因组序列进行甲基化或脱甲基化)。
干细胞可以包括全能干细胞、多潜能干细胞、多能干细胞、寡能干细胞以及单能干细胞。干细胞的具体实例包括胚胎干细胞、胎儿干细胞、成体干细胞以及诱导多潜能干细胞(ipsc)(例如参见美国公布申请号2010/0144031、2011/0076678、2011/0088107、2012/0028821,这些全部都以引用的方式并入本文)。
体细胞可以是原代细胞(非永生化细胞),如从动物中新鲜分离的细胞,或可以衍生自能够在培养中长时间增殖(例如持续长于3个月)或能够无限增殖(永生化细胞)的细胞系。成体体细胞可以从例如人类受试者的个体获得,并且根据本领域的普通技术人员可获得的标准细胞培养方案进行培养。用于本发明的方面的体细胞包括哺乳动物细胞,例如像人类细胞、非人类灵长类动物细胞或啮齿类动物(例如小鼠、大鼠)细胞。它们可以通过公知的方法从各种器官中获得,例如皮肤、肺、胰腺、肝脏、胃、肠、心脏、乳房、生殖器官、肌肉、血液、膀胱、肾脏、尿道以及其它泌尿器官等,一般从含有活体细胞的任何器官或组织获得。可用于各种实施方案中的哺乳动物体细胞包括例如成纤维细胞、塞尔托利细胞(sertolicell)、颗粒细胞、神经元、胰腺细胞、表皮细胞、上皮细胞、内皮细胞、肝细胞、毛囊细胞、角质细胞、造血细胞、黑色素细胞、软骨细胞、淋巴细胞(b淋巴细胞和t淋巴细胞)、巨噬细胞、单核白细胞、单核细胞、心肌细胞、骨骼肌细胞等。
在一些方面,一个或多个指导序列包括以位点特异性方式识别dna的序列。举例来说,指导序列可以包括crispr系统利用的指导核糖核酸(rna)序列或talen或锌指系统内以位点特异性方式识别dna的序列。所述指导序列包含与所述一个或多个基因组序列中的每一个的一部分互补的部分并且包含无催化活性的位点特异性核酸酶的结合位点。在一些实施方案中,所述rna序列被称为指导rna(grna)或单指导rna(sgrna)。
在一些方面,单个rna序列可以与正被调节或修饰的基因组序列中的一个或多个(例如全部)互补。在一个方面,单个rna与单个靶基因组序列互补。在其中要调节或修饰两个或更多个靶基因组序列的一个具体方面,引入多个(例如2个、3个、4个、5个、6个、7个、8个、9个、10个或更多个)rna序列,其中每一个rna序列与一个靶基因组序列互补(对其具有特异性)。在一些方面,两个或更多个、三个或更多个、四个或更多个、五个或更多个或六个或更多个rna序列与同一靶序列的不同部分互补(对其具有特异性)。在一个方面,两个或更多个rna序列与dna的同一区域的不同序列结合。在一些方面,单个rna序列与基因组序列的至少两个靶标或更多个靶标(例如全部)互补。对于本领域技术人员来说还将显而易见的是,rna序列中与基因组序列中的一个或多个互补的部分和rna序列中与无催化活性的位点特异性核酸酶结合的部分可以作为单个序列或作为2个(或更多个)单独的序列引入到细胞、合子、胚胎或非人类动物中。在一些实施方案中,与无催化活性的位点特异性核酸酶结合的序列包含茎-环。
在一些实施方案中,用于修饰非人类哺乳动物中的基因表达的rna序列是天然存在的rna序列、修饰的rna序列(例如包含一个或多个修饰的碱基的rna序列)、合成rna序列或其组合。如本文所用的“修饰的rna”是包含对rna序列的一个或多个修饰(例如对骨架和/或糖的修饰)的rna(例如包含一个或多个非标准和/或非天然存在的碱基的rna)。修饰rna的碱基的方法是本领域公知的。这样的修饰的碱基的实例包括核苷5-甲基胞苷(5mc)、假尿苷(ψ)、5-甲基尿苷、2'0-甲基尿苷、2-硫尿苷、n-6甲基腺苷、次黄嘌呤、二氢尿苷(d)、肌苷(i)以及7-甲基鸟苷(m7g)中所含的那些。应当指出的是,在各种实施方案中,rna序列中的任何数目的碱基可以被取代。应当进一步了解的是,可以使用不同修饰的组合。
在一些方面,所述rna序列是吗啉代寡核苷酸。吗啉代寡核苷酸通常是合成分子,具有约25个碱基的长度并且通过标准核酸碱基配对与rna的互补序列结合。吗啉代寡核苷酸具有标准核酸碱基,但是那些碱基与吗啉环而非脱氧核糖环结合,并且经由二氨基磷酸酯基而非磷酸酯连接。吗啉代寡核苷酸不会降解它们的靶rna分子,这不同于许多反义结构类型(例如硫代磷酸酯、sirna)。相反,吗啉代寡核苷酸通过空间阻断起作用并且与rna内的靶序列结合并且阻断原本可能与所述rna相互作用的分子。
每一个rna序列的长度可以从约8个碱基对(bp)到约200bp变动。在一些实施方案中,rna序列可以具有约9bp至约190bp;约10bp至约150bp;约15bp至约120bp;约20bp至约100bp;约30bp至约90bp;约40bp至约80bp;约50bp至约70bp的长度。
每一个基因组序列中与每一个rna序列互补的部分在尺寸上也可以变化。在具体方面,每一个基因组序列中与rna互补的部分可以具有约8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、31个、32个、33个、34个、35个、36个、37个、38个、39个、40个、41个、42个、43个、44个、45个、46个、47个、48个、49个、50个、51个、52个、53个、54个、55个、56个、57个、58个、59个、60个、61个、62个、63个、64个、65个、66个、67个、68个、69个、70个、71个、72个、73个、74个、75个、76个、77个、78个、79个、80个、81个、82个、83个、84个、85个、86个、87个、88个、89个、90个、81个、92个、93个、94个、95个、96个、97个、98个或100个核苷酸(连续核苷酸)的长度。在一些实施方案中,每一个rna序列可以与每一个基因组序列的部分至少约70%、75%、80%、85%、90%、95%、100%等同一或相似。在一些实施方案中,每一个rna序列与每一个基因组序列完全或部分相同或相似。举例来说,每一个rna序列可以与基因组序列的部分的完全互补序列有约1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个等核苷酸不同。在一些实施方案中,一个或多个rna序列在基因组序列的至少约10个至约25个(例如约20个)核苷酸上是完全互补的(100%)。
一个或多个指导序列(例如rna序列)可以与待修饰的靶基因组序列的多个全部或部分中的任一个互补。在一些方面,所述靶基因组序列包括差异甲基化区域、增强子(例如myod远端增强子)、启动子(例如bdnf启动子)、报告基因或ctcf结合位点。
在本发明的一些方面,所述调节一个或多个基因组序列的方法包括将与一个或多个基因组序列的(一个或多个)调节区域、开放阅读框(orf;剪接因子)、内含子序列、染色体区域(例如端粒、着丝粒)的全部或一部分互补的一个或多个指导序列引入到细胞中。在一些方面,所述基因组序列是质粒或线性双链dna(dsdna)的全部或一部分。在一些方面,由一个或多个基因组序列靶向的调节区域是启动子、增强子和/或操纵子区域。在一些方面,调节区域的全部或一部分被一个或多个基因组序列靶向。由一个或多个基因组序列靶向的区域的全部或一部分可以是差异甲基化区域。在一些方面,所述差异甲基化区域恰好在一个或多个基因(例如内源性基因;外源性基因)或(一个或多个)转录起始位点(tss)上游约25个碱基、50个碱基、100个碱基、200个碱基、300个碱基、400个碱基、500个碱基、600个碱基、700个碱基、800个碱基、900个碱基、1000个碱基、1500个碱基、2000个碱基、5000个碱基、10000个碱基、20000个碱基、50000个碱基或更多个碱基处或在上述范围内。在一些方面,所述差异甲基化区域恰好在一个或多个基因(例如内源性基因;外源性基因)或tss下游约25个碱基、50个碱基、100个碱基、200个碱基、300个碱基、400个碱基、500个碱基、600个碱基、700个碱基、800个碱基、900个碱基、1000个碱基、1500个碱基、2000个碱基、5000个碱基、10000个碱基、20000个碱基、50000个碱基或更多个碱基处或在上述范围内。如本领域的普通技术人员所将了解的那样,由一个或多个基因组序列靶向的调节区域可以完全或部分存在于基因(例如内源性或外源性)或tss的5'末端处或附近。基因的5'末端可以包括非转录(侧接)区域(例如启动子的全部或一部分)和转录区域的一部分。
如对于本领域的普通技术人员来说将显而易见的是,一个或多个rna序列还可以包含一个或多个表达控制元件。举例来说,在一些实施方案中,rna序列包含适于在细胞中引导表达的启动子,其中rna序列的一部分与一个或多个表达控制元件可操作地连接。所述启动子可以是病毒启动子(例如cmv启动子)或哺乳动物启动子(例如pgk启动子)。rna序列可以包含其它遗传元件,例如以增强转录物的表达或稳定性。在一些实施方案中,另外的编码区编码选择标记(例如报告基因,如绿色荧光蛋白(gfp))。
如本文所述,一个或多个rna序列还包含(一种或多种)无催化活性的位点特异性核酸酶的(一个或多个)结合位点。所述无催化活性的位点特异性核酸酶可以是无催化活性的crispr相关(cas)蛋白。在一个具体方面,在一个或多个rna序列与一个或多个基因组序列杂交时,所述无催化活性的位点特异性核酸酶与所述一个或多个rna序列结合。
在一些方面,所述调节一个或多个基因组序列的方法包括通过调节引入细胞或合子中的一个或多个指导序列的量(例如克、毫克、微克、纳克、摩尔、毫摩尔、微摩尔、纳摩尔、化学计量、摩尔比)来调节对一个或多个基因组序列的调节水平。在一些方面,与对相同细胞或合子中的另一个基因组序列的调节水平相比,对一个基因组序列的调节水平是相同的或不同的。在一个方面,调节多个基因组序列(例如多重化激活)。
在一个方面,所述方法还包括将一种或多种无催化活性的cas(dcas)核酸或其变体引入到细胞、胚胎、合子或非人类哺乳动物中。在一些方面,将dcas蛋白或其变体引入细胞、胚胎、合子或非人类哺乳动物中。在一些方面,细胞,例如有丝分裂后细胞、神经元、成纤维细胞、干细胞(es细胞或ips细胞)、合子、胚胎或动物可能已经携带编码dcas的核酸(可以是组成型或诱导型)和/或可能已经含有dcas蛋白。举例来说,在一些实施方案中,细胞、合子、胚胎或动物可以起源于当中已经通过涉及人工的过程引入了编码dcas蛋白的核酸的细胞或生物体。
在本发明的方法中可以使用本领域已知的多种crispr相关(cas)基因或蛋白质并且cas蛋白的选择将取决于所述方法的具体条件(例如ncbi.nlm.nih.gov/gene/?term=cas9)。cas蛋白的具体实例包括casl、cas2、cas3、cas4、cas5、cas6、cas7、cas8、cas9以及caslo。在一个具体方面,用于所述方法中的cas核酸或蛋白质是cas9。在一些实施方案中,cas蛋白,例如cas9蛋白可以来自多种原核物种中的任一种。在一些实施方案中,可以选择特定的cas蛋白,例如特定的cas9蛋白以识别特定的前间区序列邻近基序(pam)序列。在某些实施方案中,cas蛋白,例如cas9蛋白可以从细菌或古菌中获得或使用已知的方法合成。在某些实施方案中,cas蛋白可以来自革兰氏阳性细菌或革兰氏阴性细菌。在某些实施方案中,cas蛋白可以来自链球菌属(streptococcus)(例如化脓性链球菌(s.pyogenes)、嗜热链球菌(s.thermophilus))、隐球菌属(crptococcus)、棒状杆菌属(corynebacterium)、嗜血杆菌属(haemophilus)、真杆菌属(eubacterium)、巴氏杆菌属(pasteurella)、普雷沃菌属(prevotella)、韦荣球菌属(veiuonella)或海杆菌属(marinobacter)。在一些实施方案中,可以将编码两种或更多种不同的cas蛋白的核酸或两种或更多种cas蛋白引入细胞、合子、胚胎或动物中,例如以允许识别和修饰包含相同的、相似的或不同的pam基序的位点。
在一些实施方案中,所述cas蛋白是cpf1蛋白或其功能部分。在一些实施方案中,所述cas蛋白是来自任何细菌物种的cpf1或其功能部分。在某些实施方案中,cpf1蛋白是新凶手弗朗西斯氏菌(francisellanovicida)u112蛋白或其功能部分、氨基酸球菌属菌种(acidaminococcussp.)bv3l6蛋白或其功能部分或毛螺旋菌科(lachnospiraceae)细菌nd2006蛋白或其功能部分。cpf1蛋白是v型crispr系统的成员。cpf1蛋白是包含约1300个氨基酸的多肽。cpf1含有ruvc样核酸内切酶结构域。
在一些实施方案中,cas9切口酶可以通过使cas9核酸酶结构域中的一个或多个失活来产生。在一些实施方案中,cas9的ruvci结构域中的残基10处的氨基酸取代将核酸酶转化成dna切口酶。举例来说,氨基酸残基10处的天冬氨酸可以取代丙氨酸(cong等,science,339:819-823)。产生无催化活性的cas9蛋白的其它氨基酸突变包括在残基10和/或残基840处突变。残基10和残基840这两者处的突变可以产生无催化活性的cas9蛋白,其在本文有时被称为dcas9。举例来说,d10a和h840acas9突变体无催化活性。
调节一个或多个基因组序列可以包括引入一个或多个效应子结构域。如本文所用的“效应子结构域”是调节基因组序列(例如基因)的表达和/或激活的分子(例如蛋白质)。所述效应子结构域可以具有甲基化活性或脱甲基化活性(例如dna甲基化活性或dna脱甲基化活性)。在一些方面,所述效应子结构域靶向基因的一个或两个等位基因。所述效应子结构域可以作为核酸序列和/或作为蛋白质引入。在一些方面,所述效应子结构域可以是组成型或诱导型效应子结构域。在一些方面,将cas(例如dcas)核酸序列或其变体和效应子结构域核酸序列作为嵌合序列引入细胞中。在一些方面,所述效应子结构域与和cas蛋白缔合(例如结合)的分子融合(例如效应分子与结合cas蛋白的抗体或其抗原结合片段融合)。在一些方面,将cas(例如dcas)蛋白或其变体和效应子结构域融合或拴系,从而产生嵌合蛋白并且作为嵌合蛋白引入细胞中。在一些方面,cas(例如dcas)蛋白和效应子结构域通过蛋白质-蛋白质相互作用结合。在一些方面,cas(例如dcas)蛋白和效应子结构域共价连接。在一些方面,效应子结构域与cas(例如dcas)蛋白非共价缔合。在一些方面,将cas(例如dcas)核酸序列和效应子结构域核酸序列作为单独的序列和/或蛋白质引入。在一些方面,cas(例如dcas)蛋白和效应子结构域没有融合或拴系。
如本文所示,与(一个或多个)效应子结构域的全部或一部分(例如生物活性部分)拴系的无催化活性(d10a;h840a)的cas9蛋白(dcas9)的融合产生了嵌合蛋白,所述嵌合蛋白可以由一个或多个rna序列(sgrna)引导到特定的dna位点以调节一个或多个基因组序列的活性和/或表达(例如对转录或染色质组织发挥某些作用,或将特定种类的分子带入特定的dna基因座中,或用作局部组蛋白或dna状态的感受器)。在具体方面,与效应子结构域的全部或一部分拴系的dcas9的融合产生了嵌合蛋白,所述嵌合蛋白可以由一个或多个rna序列引导到特定的dna位点以调节或修饰一个或多个基因组序列的甲基化或脱甲基化。如本文所用的“效应子结构域的生物活性部分”是维持效应子结构域的功能(例如完全、部分、最低限度)的部分(例如“最小”或“核心”结构域)。cas9(例如dcas9)与一个或多个效应子结构域的全部或一部分的融合产生嵌合蛋白。
效应子结构域的实例包括转录激活结构域、共激活因子结构域、转录因子、转录暂停释放因子结构域、转录延伸负调节因子结构域、转录阻遏因子结构域、染色质组织因子结构域、重塑因子结构域、组蛋白修饰因子结构域、dna修饰结构域、rna结合结构域、蛋白质相互作用输入器结构域(grunberg和serrano,nucleicacidsresearch,3'8(8):'2663-267'5(2010))以及蛋白质相互作用输出器结构域(grunberg和serrano,nucleicacidsresearch,3'8(8):'2663-267'5(2010))。如本文所用的“蛋白质相互作用输入器”和“蛋白质相互作用输出器”指的是蛋白质-蛋白质相互作用(ppi)。在一些实施方案中,ppi可例如通过小分子或通过光来调节。在一些方面,使用无活性的cas蛋白使结合配偶体靶向到基因组中的不同位点。结合配偶体相互作用,从而使靶向的基因座接近。蛋白质相互作用输出器是用于检测/监测ppi的发生的系统,一般通过当发生ppi时产生可检测的信号(例如通过重构荧光蛋白)或触发特定细胞响应(例如通过重构胱天蛋白酶蛋白以诱导细胞凋亡)。在这种背景下的构思是用“输出器”的组分靶向基因组中的不同位点。如果发生相互作用,那么“输出器”产生信号。这可以用于确定或监测靶向的基因座的接近程度。在一些方面,用试剂处理细胞并且确定所述试剂对细胞的作用。效应子结构域的其它实例包括组蛋白标记识别因子(reader)/相互作用因子(cell.com/abstract/s0092-8674(10)00951-7)和dna修饰识别因子/相互作用因子。
在一些方面,所述效应子结构域是dna修饰因子。dna修饰因子的具体实例包括从5mc转化成5hmc,如tetl(tetlcd);通过tetl、acida、mbd4、apobecl、apobec2、apobec3、tdg、gadd45a、gadd45b、ros1进行dna脱甲基化;通过dnmtl、dnmt3a、dnmt3b、cpg甲基转移酶m.sssi和/或m.ecohk31i进行dna甲基化。在具体方面,效应子结构域是tet1。在其它具体方面,效应子结构域是dmnt3a。在一些实施方案中,dcas9与tet1融合。在其它实施方案中,dcas9与dnmt3a融合。
dna甲基化通过两种从头dna甲基转移酶(dnmt3a/b)建立,并且通过dnmt1维持(smith和meissner,2013)。在发育期间的基因激活与启动子和增强子序列的脱甲基化有关。此外,脱甲基化可以通过tet(10-11易位)双加氧酶氧化甲基以形成5-羟甲基胞嘧啶(5-hmc),然后通过dna复制依赖性稀释或dna糖基化酶引发的碱基切除修复(ber)恢复成未修饰的胞嘧啶来实现,该过程被称作主动脱甲基化并且被提出在特定发育阶段期间,如植入前胚胎或有丝分裂后神经元中起作用(wu和zhang,2014)。
在本发明的一个方面,dcas9与效应子结构域的融合可以是与全长或部分长度的效应子的单个拷贝或多个拷贝/串联拷贝的融合。其它融合可以是与效应子结构域的拆分(功能互补)型式融合。用于所述方法中的效应子结构域包括以下类别的蛋白质中的任一种:介导dna的药物诱导性成环和/或基因组基因座的接触的蛋白质、有助于由dcas9和不同的sgrna结合的基因组基因座的三维接近的蛋白质。
效应子结构域的其它实例描述于pct申请号pct/us2014/034387和美国申请号14/785031中,这些参考文献以引用的方式整体并入本文。
在一些方面,本发明涉及编码包含与效应子结构域的全部或一部分融合的cas(例如dcas)蛋白的全部或一部分的融合蛋白(嵌合蛋白)的核酸序列(例如包含所述核酸序列、基本上由所述核酸序列组成、由所述核酸序列组成的组合物)。在一些方面,本发明涉及包含与效应子结构域的全部或一部分融合的cas(例如dcas)蛋白的全部或一部分的融合蛋白(例如包含所述融合蛋白、基本上由所述融合蛋白组成、由所述融合蛋白组成的组合物)。在一些方面,cas(例如dcas)蛋白的全部或一部分靶向而不切割核酸序列。在一些方面,cas(例如dcas)蛋白可以与效应子结构域的n末端或c末端融合。在一些方面,效应子结构域的一部分调节基因组序列的甲基化(例如将基因组序列进行脱甲基化或甲基化)。
在一些方面,编码所述融合蛋白的核酸序列和/或所述融合蛋白是分离的。如本文所用的“分离的”、“基本上纯的”或“基本上纯的和分离的”核酸序列是与通常侧接所述基因或核苷酸序列的核酸(如在基因组序列中)分离和/或已经完全或部分从其它转录序列(例如在rna文库或cdna文库中)纯化的核酸序列。举例来说,本发明的分离的核酸相对于其中它天然存在的复杂细胞环境或当通过重组技术产生时的培养基或当化学合成时的化学前体或其它化学品可以是基本上分离的。如本文所用的“分离的”、“基本上纯的”或“基本上纯的和分离的”蛋白质(例如嵌合蛋白;融合蛋白)是从以下中分离或相对于以下基本上分离的蛋白质:其中它天然存在的复杂细胞环境;或当通过重组技术产生时的培养基;或当化学合成时的化学前体或其它化学品。在一些情况下,分离的材料将形成组合物(例如含有其它物质的粗提物)、缓冲系统或试剂混合物的一部分。在其它情况下,所述材料可以被纯化到必要的均一性,例如通过琼脂糖凝胶电泳或柱色谱,如hplc所测定。优选的是,分离的核酸分子占存在的所有大分子物质的至少约50%、80%、90%、95%、98%或99%(以摩尔为基础)。
在一个方面,cas9与一个或多个效应子结构域的全部或一部分的融合体包含一个或多个接头。如本文所用的“接头”是将两个或更多个效应子结构域连接或融合的物质(例如参见hermanson,bioconjugatetechniques,第2版,其在此以引用的方式整体并入本文)。如本领域的普通技术人员所将了解的那样,可以使用多种接头。在一个方面,接头包含一个或多个氨基酸。在一些方面,接头包含两个或更多个氨基酸。在一个方面,接头包含氨基酸序列gs。在一些方面,cas9(例如dcas9)与两个或更多个效应子结构域的融合体在所述结构域之间包含一个或多个散布的接头(例如gs接头)。在一些方面,一个或多个核定位序列可以位于无催化活性的核酸酶(例如dcas9)与效应子结构域之间。举例来说,融合蛋白可以包括dcas9-nls-tet1或dcas9-nls-dnmt3a。
在本发明的一些方面,所述调节细胞中的一个或多个基因组序列的方法还可以包括引入效应分子。如本文所用的“效应分子”是与效应子结构域缔合(例如结合;特异性结合)以调节基因组序列的甲基化或脱甲基化的分子(例如核酸序列;蛋白质;有机分子;无机分子、小分子)或物理触发剂(例如诱导分子;触发分子)。可以使效应分子与细胞接触和/或引入细胞中(例如作为核酸序列或作为蛋白质序列)。在一些实施方案中,所述效应分子是内源性的。在其它实施方案中,所述效应分子是外源性的。举例来说,可以将外源性效应分子引入细胞中。在一些方面,所述效应分子与效应子结构域结合。在一些方面,所述效应分子是核酸、蛋白质、药物、小有机分子以及其衍生物/变体。在本发明的一些方面,所述效应分子是抗生素或其衍生物/变体。
如对于本领域技术人员来说将显而易见的是,所述方法还可以包括将其它分子或因子引入到细胞中以促进基因组序列的甲基化或脱甲基化。抑制或增强dna甲基化的试剂可以是内源性dna甲基化酶或dna脱甲基酶的抑制剂。举例来说,dna甲基化抑制剂可以是小分子,例如胞苷类似物,如5-氮杂胞苷(阿扎胞苷(azacitidine))和5-氮杂脱氧胞苷(地西他滨(decitabine))。在其它方法中,可以向个体施用抑制或增强dna甲基化的试剂。
多个基因组序列可以使用本文所述的方法来调节或修饰并且将取决于所期望的结果。在一个方面,靶基因组序列是基因序列。在具体方面,本文所述的方法可以用于遗传修饰同一基因家族中的两个或更多个不同的基因;具有冗余功能的两个或更多个基因(例如冗余可以意指需要使所述基因中的至少两个失活以产生特定表型,例如可检测的表型);具有至少80%、85%、90%、95%、96%、97%、98%、99%或更大的同一性的两个或更多个基因;相同基因的两个或更多个拷贝;同一生物途径(例如信号传导途径、代谢途径)中的两个或更多个基因;共有至少一种生物活性和/或作用于至少一种共同底物和/或作为同一蛋白质或蛋白质-核酸复合物(例如异寡聚蛋白、剪接体、蛋白酶体、risc、转录复合物、复制复合物、着丝点、通道、转运蛋白)的一部分的两个或更多个基因。在一些方面,两个或更多个指导序列可以将包含与效应子结构域融合的无催化活性的位点特异性核酸酶的多肽引导到位于基因组序列内的不同位点。
“调节”或“修饰”与它在本领域中的用途一致地使用,即意指在所关注的过程、途径或现象中引起或促进质变或量变、改变或修饰。在没有限制的情况下,这样的变化可以是所述过程、途径或现象的不同组分或分支的相对强度或活性的增加、减少或变化。“调节剂”或“修饰剂”是在所关注的过程、途径或现象中引起或促进质变或量变、改变或修饰的试剂。
在一些方面,“调节”或“修饰”基因组序列的甲基化指的是对一个或多个基因组序列的甲基化状态的多种改变中的任一种。举例来说,所述调节一个或多个基因组序列的甲基化的方法包括将所述基因组序列进行甲基化或脱甲基化(例如可以将所述基因组序列甲基化或可以将所述基因组序列脱甲基化)。
本文提供的方法还可以用于修饰或调节细胞组合物中存在的细胞中的一个或多个基因组序列,如胚胎、合子、胎儿以及出生后哺乳动物。在一些实施方案中,在进行本文所述的方法之前,细胞(例如有丝分裂后细胞、神经元、成纤维细胞、干细胞等)、合子、胚胎或出生后哺乳动物已经被遗传修饰(已经携带一个或多个遗传修饰,例如表观遗传修饰)。举例来说,所述细胞、合子、胚胎或出生后哺乳动物可以是当中已经通过涉及人工的过程引入了外源性核酸的细胞、合子、胚胎或出生后哺乳动物(或可以至少部分地起源于当中已经通过涉及人工的过程引入了外源性核酸的细胞或生物体)。所述核酸可以例如含有所述细胞外源性的序列,它可以含有天然序列(即天然存在于细胞中的序列),但是呈非天然存在的排列(例如与来自不同基因的启动子连接的编码区)或改变型式的天然序列等。在一些实施方案中,在进行本文所述的方法之前,细胞、合子、胚胎或出生后哺乳动物尚未被遗传修饰(尚未携带一个或多个遗传修饰)。
在一些方面,本发明涉及一种产生在一个或多个基因组序列中携带修饰的非人类哺乳动物的方法,所述方法包括向合子或胚胎中引入与具有甲基化活性或脱甲基化活性的效应子结构域融合的无催化活性的位点特异性核酸酶和一个或多个指导序列。将所述合子或所述胚胎维持在以下条件下,其中所述指导序列与所述一个或多个基因组序列中的每一个的一部分杂交,并且所述与效应子结构域融合的无催化活性的位点特异性核酸酶将所述基因组序列进行甲基化或脱甲基化,从而产生具有一个或多个修饰的基因组序列的胚胎。可以将所述具有一个或多个修饰的基因组序列的胚胎转移到代孕非人类哺乳动物母体中。将所述代孕非人类哺乳动物母体维持在其中产生携带所述一个或多个修饰的基因组序列的一个或多个后代的条件下,从而产生在一个或多个基因组序列中携带修饰的非人类哺乳动物。
如对于本领域技术人员来说将显而易见的是,非人类哺乳动物也可以使用本文所述的方法和/或使用常规方法来产生,参见例如美国公布申请号2011/0302665。产生非人类哺乳动物胚胎的方法可以包括将非人类哺乳动物es细胞(例如ipsc)注射到非人类四倍体胚泡中并且将所述所得的四倍体胚泡维持在引起胚胎形成的条件下,从而产生非人类哺乳动物胚胎。在一些实施方案中,所述非人类哺乳动物细胞是小鼠细胞并且所述非人类哺乳动物胚胎是小鼠。在一些实施方案中,所述小鼠细胞是突变型小鼠细胞并且通过显微注射注射到所述非人类四倍体胚泡中。在一些实施方案中,使用激光辅助显微操作或压电注射。在一些实施方案中,非人类哺乳动物胚胎包括小鼠胚胎。
这样的常规技术的另一个实例是两步克隆,其包括将包含一个或多个突变的胚胎干(es)细胞和/或诱导多潜能干(ips)细胞引入到胚泡(例如四倍体胚泡)中并且将所述胚泡维持在引起胚胎发育的条件下。然后将所述胚胎转移(植入)到适当的代孕母体中,如假孕雌性(例如与胚胎相同的物种)。然后将所述代孕母体维持在引起携带所述一个或多个突变的活后代发育的条件下。
另一个实例是使用四倍体补偿测定,其中将两个哺乳动物胚胎的细胞组合以形成新的胚胎(tarn和rossant,develop,750:6156-6163(2003))。所述测定包括产生四倍体细胞,其中每一个染色体存在四倍。这是通过在两细胞期获取胚胎并且通过施加电流使两个细胞融合来完成的。所得的四倍体细胞继续分裂并且所有子细胞也将是四倍体。这样的四倍体胚胎正常发育到胚泡期并且将植入子宫壁中。在四倍体补偿测定中,将四倍体胚胎(处于桑椹胚或胚泡期)与来自不同生物体的正常二倍体胚胎干细胞(es)组合。胚胎正常发育;胎儿仅衍生自es细胞,而胚胎外组织仅衍生自四倍体细胞。
用于产生非人类哺乳动物的另一种常规方法包括原核显微注射。在受精后立即将dna直接引入到非人类哺乳动物卵细胞的雄性原核中。类似于上述两步克隆,将卵细胞植入到假孕雌性体内。针对整合的转基因筛选后代。随后可以使杂合后代交配以产生纯合动物。
多种非人类哺乳动物可以用于本文所述的方法中。举例来说,所述非人类哺乳动物可以是啮齿类动物(例如小鼠、大鼠、豚鼠、仓鼠)、非人类灵长类动物、犬、猫、牛、马、猪或羊。
在一些方面,各种小鼠品系和人类疾病的小鼠模型与本文所述的产生在一个或多个靶核酸序列中携带突变的非人类哺乳动物的方法结合使用。本领域的普通技术人员了解用于对人类疾病进行建模的实验室小鼠的数千种可商购获得和非可商购获得的品系。存在用于诸如癌症、心血管疾病、自身免疫性疾病和病症、炎症性疾病、糖尿病(1型和2型)、神经疾病以及其它疾病的疾病的小鼠模型。可商购获得的研究品系的实例包括而不限于11bhsd2小鼠、gsk3b小鼠、129-e小鼠、hsd11b1小鼠、ak小鼠
在一些方面,所述产生在一个或多个基因组序列中携带修饰的非人类哺乳动物的方法还包括使一种或多种可商购获得和/或非可商购获得的非人类哺乳动物与通过本文所述的方法产生的在一个或多个基因组序列中携带修饰的非人类哺乳动物交配。本发明还涉及通过本文所述的方法产生的非人类哺乳动物。
在一些方面,基因组序列与疾病或疾患有关(例如参见vanderweyden等,genomebiol,12:224(2011))。所关注的遗传修饰的具体实例包括修饰一个或多个序列(例如一个或多个基因)以匹配不同物种中的序列(例如对于所关注的任何一个或多个基因,将小鼠序列变为人类序列);改变潜在或已知的蛋白质翻译后修饰位点(例如磷酸化、糖基化、脂质化、酰化、乙酰化);改变潜在或已知的表观遗传修饰位点;改变潜在或已知的蛋白质-蛋白质或蛋白质-核酸相互作用位点;插入标签,例如表位标签;和/或插入剪接位点或使剪接位点缺失。
在一些方面,修饰一个或多个基因组序列的一个拷贝。在一些方面,修饰细胞中基因组序列中的一个或多个的两个拷贝。在一些方面,所修饰的一个或多个基因组序列是细胞内源性的。
在具体方面,所述基因组序列中的至少两个是内源性基因组序列。在一些方面,所述基因组序列中的至少两个是外源性基因组序列。在其中存在至少两个基因组序列的一些方面,所述基因组序列中的至少一个是内源性基因组序列并且所述基因组序列中的至少一个是外源性基因组序列。在一些方面,所述基因组序列中的至少两个是内源性基因。在一些方面,所述基因组序列中的至少两个是外源性基因。在其中存在至少两个基因组序列的一些方面,所述基因组序列中的至少一个是内源性基因并且所述基因组序列中的至少一个是外源性基因。在一些方面,所述基因组序列中的至少两个相隔至少1kb。在一些方面,所述基因组序列中的至少两个在不同的染色体上。基因组序列可以包含标签(例如表位标签或荧光标签)或转基因(例如报告基因)。
本文提供的方法提供了细胞、胚胎、合子以及非人类哺乳动物中的多重化基因组编辑。如本文所示,可以在单个步骤中产生在多个基因中携带修饰的细胞、胚胎、合子以及非人类哺乳动物。在一些方面,本文所述的方法允许使用本文所述的方法修饰(单个)细胞、合子、胚胎或非人类哺乳动物中的1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个等基因组序列(例如基因)。在一个具体方面,在(单个)细胞、合子、胚胎或非人类哺乳动物中修饰一个基因组序列。在一些方面,在(单个)细胞、合子、胚胎或非人类哺乳动物中修饰两个基因组序列。在一些方面,在(单个)细胞、合子、胚胎或非人类哺乳动物中修饰三个基因组序列。在一些方面,在(单个)细胞、合子、胚胎或非人类哺乳动物中修饰四个基因组序列。在一些方面,在(单个)细胞、合子、胚胎或非人类哺乳动物中修饰五个基因组序列等。
如对于本领域技术人员来说将显而易见的是,可以使用多种方法将核酸和/或蛋白质引入到细胞、合子、胚胎和/或哺乳动物中。合适的方法包括磷酸钙或脂质介导的转染、电穿孔、注射以及使用载体(例如病毒载体,如腺病毒载体)转导或感染。在一些方面,将核酸和/或蛋白质与运载体,例如阳离子运载体复合,所述运载体促进核酸和/或蛋白质的摄取(例如经由内吞)。
本文所述的方法还可以包括分离通过所述方法产生的细胞或合子。因此,在一些方面,本发明涉及通过本文所述的方法产生的细胞或合子(分离的细胞或合子)。在一些方面,本公开提供了携带一个或多个修饰的细胞的克隆群体、复制包含携带一个或多个修饰的细胞的培养物以及从所产生的动物中分离的细胞。
本文所述的方法还可以包括使所产生的动物与携带遗传修饰(任选地在相同的品系背景下)和/或具有所关注的一个或多个表型(例如疾病易感性,如nod小鼠)的其它动物杂交。此外,所述方法可以包括修饰来自携带一个或多个遗传修饰和/或具有所关注的一个或多个表型(例如疾病易感性)的品系的细胞、合子和/或动物。在一些方面,所述遗传修饰是表观遗传修饰。
本文所述的方法还可以包括使用多种已知的方法评估一个或多个靶核酸是否已经被修饰和/或调节。
在一些实施方案中,本文所述的方法用于在细胞、合子、胚胎或动物中产生多个遗传修饰,其中所述遗传修饰中的至少一个将基因进行甲基化或脱甲基化,并且所述遗传修饰中的至少一个在不同的基因或基因组位置中。在一些实施方案中,遗传修饰还包括表观遗传修饰。分析所得的细胞、合子、胚胎或动物或由其产生的细胞、合子、胚胎或动物。在一些实施方案中,所述遗传修饰中的至少一个可以是条件性的(例如所述修饰的作用,如基因甲基化或脱甲基化仅在某些条件下变得明显,这通常在技术人员的控制下)。在一些实施方案中,允许动物至少发育到出生后阶段,例如成年阶段。施加使修饰产生作用的适当条件(有时被称作“诱导条件”),并且随后分析动物的表型。可以将表型与未修饰的动物的表型或在施加诱导条件之前的表型进行比较。
分析可以包括本领域已知的任何类型的表型分析,例如研究任何组织、器官或器官系统(或整个生物体)的结构、尺寸、发育、重量或功能、分析行为、任何生物途径或过程的活动、任何特定物质或基因产物的水平等。在一些实施方案中,分析包括基因表达分析,例如在mrna或蛋白质的水平上。在一些实施方案中,这样的分析可以包括例如使用微阵列(例如寡核苷酸微阵列,有时被称作“芯片”)、高通量测序(例如rnaseq)、chiponchip分析、chlpseq分析等。在一些实施方案中,可以使用高内涵筛选,其中高通量筛选的元件可以经由使用自动化显微镜术和图像分析应用于分析单个细胞(参见例如zanella等,(2010).highcontentscreening:seeingisbelieving.trendsbiotechnol.28:237-245)。在一些实施方案中,分析包括定量分析细胞的组分,如单个蛋白质的时空分布、细胞骨架结构、囊泡以及细胞器,例如在与测试剂,例如化合物接触时。在一些实施方案中,可以评估单个蛋白质和蛋白质-蛋白质相互作用的激活或抑制和/或生物过程和细胞功能的变化。用于生物过程、功能以及细胞组分的一系列荧光探针是可获得的并且可以例如与荧光显微镜术一起使用。在一些实施方案中,根据本文的方法产生的细胞或动物可以包含报告基因,例如荧光报告基因或荧光酶(例如荧光素酶,如高斯(gaussia)荧光素酶、海肾荧光素酶或萤火虫荧光素酶),其例如报告特定基因的表达或活性。这样的报告基因可以与蛋白质融合,从而使得所述蛋白质或它的活性是可检测的,任选地使用非侵入性检测手段,例如成像或诸如pet成像、mri、荧光检测的检测手段。根据本发明的多重化基因组编辑可以允许安装报告基因以检测例如细胞、组织、器官或动物,例如活体动物中的多种蛋白质,例如2-20种不同的蛋白质。
根据本发明的多重化基因组编辑可以用于确定或研究具有未知功能的基因的一种或多种生物学作用和/或在疾病中的作用。举例来说,发现由第一基因和第二基因中的修饰所引起的合成效应可以精确定位其中涉及所述一个或多个基因或一种或多种编码的基因产物的遗传或生化途径。
在一些实施方案中,考虑在本文所述的方法中使用在诸如国际基因敲除小鼠联盟(internationalknockoutmouseconsortium,ikmc)(其网站是knockoutmouse.org)的项目中产生的动物中产生或源自于所述动物的细胞或合子。在一些实施方案中,考虑使如本文所述产生的动物与由ikmc产生或经由ikmc获得的动物杂交。举例来说,在一些实施方案中,待根据本文所述的方法修饰的小鼠基因是来自可获得序列和基因组坐标的小鼠基因组信息学(mousegenomeinformatics,mgi)数据库的任何基因,例如由ncbi、ensembl以及vega(脊椎动物基因组注释(vertebrategenomeannotation))流水线对于小鼠基因组版本37(mousegenomebuild37)(ncbi)或基因组参照序列联盟(genomereferenceconsortium)grcm38预测的任何基因。
在一些实施方案中,待修饰的基因或基因组位置被包括在存在完全测序的基因组的物种的基因组中。基因组序列可以例如从ucsc基因组浏览器(ucscgenomebrowser)(genome.ucsc.edu/index.html)获得。举例来说,在一些实施方案中,待根据本文所述的方法修饰的人类基因或序列可以见于人类基因组版本hgl9(基因组参照序列联盟)中。在一些实施方案中,基因是在ncbi的基因数据库(ncbi.nlm.nih.gov/gene)中已经指定了基因id的任何基因。在一些实施方案中,基因是可在数据库中获得基因组、cdna、mrna或编码的基因产物(例如蛋白质)序列的任何基因,如可在国家生物技术信息中心(ncbi.nih.gov)或通用蛋白质资源(universalproteinresource)(uniprot.org)获得的那些中的任一种。数据库包括例如基因库、refseq、gene、uniprotkb/swissprot、uniprotkb/trembl等。
在一些实施方案中,关注的是,对已知或疑似的差异甲基化区域(dmr)进行遗传修饰。存在差异甲基化区域的各种实例。差异甲基化区域可以在不同细胞类型的细胞(例如肌细胞与神经元或皮肤细胞与肝细胞)之间差异甲基化。差异甲基化区域也可以在患病细胞与非患病细胞(例如癌细胞与非癌细胞)之间差异甲基化。差异甲基化区域也可以在分化状态(例如祖细胞与终末分化细胞)之间差异甲基化。可以评估对一个或多个基因(例如与所述修饰相距最多约0.5kb、1kb、2kb、5kb、10kb、20kb、50kb、100kb、500kb以内或约1mb、2mb、5mb或10mb以内)的表达的影响。可以在序列中进行遗传修饰以确定这样的遗传修饰是否改变细胞或动物的表型或影响rna或蛋白质的产物或改变对疾病的易感性。遗传修饰可以包括表观遗传修饰。在一些方面,差异甲基化区域可以是过度甲基化的或非甲基化的。
在一些方面,关注的是,将异常过度甲基化的基因组序列脱甲基化或将异常非甲基化的基因组序列甲基化。在一些方面,异常过度甲基化的序列或异常非甲基化的序列可以存在于疾病或病症中。在其它方面,关注的是,将异常非甲基化的ctcf位点(例如ctcf结合位点)甲基化或去除异常甲基化的ctcf位点的甲基化。修饰ctcf位点的甲基化或脱甲基化可以治疗或预防表现出异常非甲基化的序列或区域或异常过度甲基化的序列或区域的疾病或病症。举例来说,如果希望增加基因的表达(例如如果所述基因的表达异常低和/或如果期望增加表达以实现治疗或其它目的),那么可以通过将ctcf结合位点甲基化来打开ctcf环,从而使所述在所述环之外的基因处于所述环内部的增强子的控制之下。
在一些方面,本文所述的方法可以用于产生在启动子序列中具有修饰的细胞。使dcas9-tet1融合蛋白或dcas9-dnmt3a融合蛋白靶向到甲基化的启动子序列或非甲基化的启动子序列分别引起内源性报告基因的激活或沉默。举例来说,dcas9-tet1融合蛋白靶向bdnf启动子iv并且将所述启动子脱甲基化,从而诱导有丝分裂后神经元中的bdnf表达。
在一些方面,本文所述的方法可以用于产生在增强子序列中具有修饰的细胞。使dcas9-tet1融合蛋白或dcas9-dnmt3a融合蛋白靶向到甲基化的增强子序列或非甲基化的增强子序列分别引起内源性增强子的激活或沉默。举例来说,dcas9-tet1融合蛋白靶向成纤维细胞中的myod远端增强子并且将所述增强子脱甲基化,从而促进成纤维细胞重编程为成肌细胞。
在其它方面,本文所述的方法可以用于产生在ctcf结合位点中具有修饰的细胞。使dcas9-tet1融合蛋白或dcas9-dnmt3a融合蛋白靶向到ctcf结合位点可以影响ctcf结合并且干扰dna成环。举例来说,dcas9-dnmt3a融合蛋白对ctcf环锚定位点进行靶向从头甲基化,阻断ctcf结合并且干扰dna成环,从而引起相邻环中的基因表达改变。
在一些实施方案中,本文所述的任何方法可以包括从根据本文所述的方法产生的动物,例如,如本文所述产生的任何遗传修饰的动物中分离一种或多种细胞、样品或物质。在一些实施方案中,方法还可以包括分析所述一种或多种细胞、样品或物质。这样的分析可以例如评估根据所述方法引入的一个或多个遗传修饰的作用。遗传修饰可以包括基因组序列的甲基化或脱甲基化和/或可以包括表观遗传修饰。
在一些实施方案中,根据本文所述的方法产生的动物可以用于鉴定用于治疗疾病的候选药剂和/或测试药剂的潜在毒性或副作用。在一些实施方案中,本文所述的任何方法可以包括使根据本文所述的方法产生的动物,例如,如本文所述产生的任何遗传修饰的动物与测试剂(例如小分子、核酸、多肽、脂质等)接触。在一些实施方案中,接触包括施用所述测试剂。施用可以通过任何途径(例如口服、静脉内、腹膜内、管饲、局部、透皮、肌内、肠内、皮下)进行,可以是全身的或局部的,可以包括任何剂量(例如约0.01mg/kg至约500mg/kg),可以包括单次剂量或多次剂量。在一些实施方案中,方法还可以包括分析所述动物。这样的分析可以例如评估测试剂在具有根据所述方法引入的一个或多个遗传修饰的动物中的作用。在一些实施方案中,可以鉴定降低或增强一个或多个遗传修饰的作用的测试剂。在一些实施方案中,如果测试剂减少或抑制与一个或多个遗传修饰相关或由一个或多个遗传修饰产生的疾病的发生(或减少或抑制这样的疾病的一个或多个症状或体征),那么所述测试剂可以被鉴定为用于治疗与所述一个或多个遗传修饰相关或由所述一个或多个遗传修饰产生或与携带所述遗传修饰的基因或基因组位置中天然存在的突变相关或由所述天然存在的突变产生的疾病的候选药剂。
在一些实施方案中,细胞可以是患病细胞或可以源自于患有疾病,例如影响所述细胞或用于获得所述细胞的器官的疾病的受试者。在一些实施方案中,将突变引入细胞的与疾病(例如所关注的任何疾病,如本文提到的疾病)有关的基因组区域中。举例来说,在一些实施方案中,关注的是,将已知或疑似参与疾病发病机制和/或已知或疑似与增加或降低发生疾病或疾病的一个或多个特定表现的风险有关的基因或基因组位置甲基化或脱甲基化。在一些实施方案中,关注的是,将基因或基因组位置甲基化或脱甲基化,并且确定这样的修饰是否改变发生疾病或疾病的一个或多个表现的风险、改变疾病的进展或改变受试者对疾病的治疗或候选治疗的响应。在一些实施方案中,关注的是,将异常的或疾病相关的核苷酸或序列修饰成正常的或与疾病无关的核苷酸或序列。在一些实施方案中,这可以允许产生仅在一个或多个所选择的遗传修饰位点处不同的遗传匹配的细胞或细胞系(例如ips细胞或细胞系)。如本文所述的多重化基因组编辑可以允许产生除了例如2个至20个所选择的遗传改变位点之外,同基因的细胞或细胞系。这可以允许研究疑似或已知在疾病风险、发生或进展中起作用的多个修饰的组合作用。
术语“疾病”、“病症”或“疾患”是可互换使用的并且可以指的是从生物体的健康和/或发挥正常功能的状态的任何改变,例如对患病个体造成疼痛、不适、功能障碍、痛苦、退化或死亡的身体或精神的异常。疾病包括本领域的普通技术人员已知的任何疾病。在一些实施方案中,疾病是慢性疾病,例如,它通常持续或已经持续至少3-6个月或更长时间,例如1年、2年、3年、5年、10年或更多年或无限期。疾病可以具有在患有所述疾病的个体中通常发生的一组特征性的症状和/或体征。疾病以及其诊断和治疗方法描述于标准医学教科书,如longo,d.等(编著),harrison'sprinciplesofinternalmedicine,第18版;mcgraw-hillprofessional,2011和/或goldman'scecilmedicine,saunders;第24版(2011年8月5日)中。在某些实施方案中,疾病是多基因病症(也被称为复杂、多因子或多基因病症)。这样的疾病可能与多个基因的作用相关,有时与环境因素(例如暴露于特定的物理剂或化学剂或生物剂(如病毒)、生活方式因素,如饮食、吸烟等)组合。多基因病症可以是其中已知或怀疑多个基因(例如这些基因的特定等位基因、这些基因中的特定多态性)可以促进发生所述疾病的风险和/或可以促进所述疾病表现的方式(例如它的严重程度、发病年龄、进展速率等)的任何疾病。在一些实施方案中,多基因疾病是具有如由家族聚集性(在某些家族中比在一般群体中更常发生)所示的遗传组分,但是不遵循孟德尔遗传定律(mendelianlawsofinheritance)的疾病,例如,所述疾病不明显遵循显性、隐性、x连锁或y连锁遗传模式。在一些实施方案中,多基因疾病是通常不受单个基因中的大效应变体控制(在孟德尔病症(mendeliandisorder)的情况下往往如此)的疾病。在一些实施方案中,多基因疾病可以家族形式和散发形式发生。实例包括例如帕金森氏病(parkinson'sdisease)、阿尔茨海默氏病(alzheimer'sdisease)以及各种类型的癌症。多基因疾病的实例包括许多常见疾病,如高血压、糖尿病(例如ii型糖尿病)、心血管疾病、癌症以及中风(缺血性、出血性)。在一些实施方案中,疾病,例如多基因疾病是精神疾病、神经疾病、神经发育疾病、神经退行性疾病、心血管疾病、自身免疫性疾病、癌症、代谢疾病或呼吸系统疾病。在一些实施方案中,至少一个基因牵涉到多基因疾病的家族形式。
在一些实施方案中,疾病是癌症,该术语一般可互换地用于指以一个或多个肿瘤,例如一个或多个恶性或潜在恶性肿瘤为特征的疾病。如本文所用的术语“肿瘤”包括异常生长,其包括异常增殖细胞。如本领域已知的那样,肿瘤通常以未受到适当调节的过度细胞增殖为特征(例如其对通常将限制增殖的生理影响和信号没有正常响应)并且可以表现出以下特性中的一种或多种:发育不良(例如缺乏正常细胞分化,从而导致未成熟细胞的数量或比例增加);间变性(例如更大的分化丧失、更多的结构组织丧失、细胞多形性、异常,如大而深染的核、高核/细胞质比、非典型有丝分裂等);侵袭邻近组织(例如破坏基底膜);和/或转移。恶性肿瘤具有持续生长的趋势和扩散,例如局部侵袭和/或局部和/或向远处位置转移的能力,而良性肿瘤常常保留在起源部位并且在生长方面常常是自限性的。术语“肿瘤”包括恶性实体肿瘤,例如癌瘤(由上皮细胞产生的癌症)、肉瘤(由间充质来源的细胞产生的癌症)以及恶性生长,其中可能不存在可检测的实体肿瘤块(例如某些血液恶性肿瘤)。癌症包括但不限于:乳腺癌;胆道癌;膀胱癌;脑癌(例如成胶质细胞瘤、成神经管细胞瘤);宫颈癌;绒毛膜癌;结肠癌;子宫内膜癌;食道癌;胃癌;血液赘生物,包括急性淋巴细胞性白血病和急性骨髓性白血病;t细胞急性成淋巴细胞性白血病/淋巴瘤;毛细胞白血病;慢性淋巴细胞性白血病、慢性骨髓性白血病、多发性骨髓瘤;成人t细胞白血病/淋巴瘤;上皮内赘生物,包括鲍文病(bowen'sdisease)和佩吉特氏病(paget'sdisease);肝癌;肺癌;淋巴瘤,包括霍奇金氏病(hodgkin'sdisease)和淋巴细胞性淋巴瘤;成神经细胞瘤;黑色素瘤;口腔癌,包括鳞状细胞癌;卵巢癌,包括由上皮细胞、基质细胞、生殖细胞以及间充质细胞产生的卵巢癌;成神经细胞瘤、胰腺癌;前列腺癌;直肠癌;肉瘤,包括血管肉瘤、胃肠道间质肿瘤、平滑肌肉瘤、横纹肌肉瘤、脂肪肉瘤、纤维肉瘤以及骨肉瘤;肾癌,包括肾细胞癌和肾母细胞瘤(wilmstumor);皮肤癌,包括基底细胞癌和鳞状细胞癌;睾丸癌,包括生殖肿瘤,如精原细胞瘤、非精原细胞瘤(畸胎瘤、绒毛膜癌)、间质肿瘤以及生殖细胞肿瘤;甲状腺癌,包括甲状腺腺癌和髓样癌。应当了解的是,多种不同的肿瘤类型可以在某些器官中出现,其在例如临床和/或病理特征和/或分子标志物方面可以不同。在多种不同的器官中出现的肿瘤被论述,例如who肿瘤分类系列(whoclassificationoftumoursseries)(第4版或第3版(肿瘤病理学和遗传学系列(pathologyandgeneticsoftumoursseries)),国际癌症研究机构(internationalagencyforresearchoncancer,iarc),瑞士日内瓦的who出版社(whopress,geneva,switzerland)),其所有卷都以引用的方式并入本文。在一些实施方案中,癌症是其中特定基因的突变或过表达已知或疑似在癌症的发生、进展、复发等中起作用的癌症。在一些实施方案中,这些基因是根据本文所述的方法进行遗传修饰的靶标。在一些实施方案中,基因是癌基因、原癌基因或肿瘤抑制基因。术语“癌基因”包括在表达时可以增加癌症发生或进展的可能性或导致癌症发生或进展的核酸。正常细胞序列(“原癌基因”)可以通过突变和/或异常表达激活以变成癌基因(有时被称作“激活的癌基因”)。在各种实施方案中,癌基因可以包含基因产物的完整编码序列或至少部分地维持完整序列的致癌潜力的部分或编码融合蛋白的序列。致癌突变可以导致例如蛋白质活性改变(例如增加)、适当调节的丧失或ra或蛋白质水平的改变(例如增加)。异常表达可能例如由于导致与诸如增强子的调节元件的并置的染色体重排、表观遗传机制或由于扩增而发生,并且可能导致原癌基因产物的量增加或在不适当的细胞类型中产生。原癌基因常常编码控制或参与细胞增殖、分化和/或细胞凋亡的蛋白质。这些蛋白质包括例如各种转录因子、染色质重塑因子、生长因子、生长因子受体、信号转导蛋白以及细胞凋亡调节因子。tsg可以是任何基因,其中所述基因的表达产物的功能的丧失或降低可以增加癌症发生或进展的可能性或导致癌症发生或进展。功能的丧失或降低可以例如由于突变或表观遗传机制而发生。许多tsg编码通常用以限制或负调节细胞增殖和/或促进细胞凋亡的蛋白质。示例性癌基因包括例如myc、src、fos、jun、myb、ras、raf、abl、alk、akt、trk、bcl2、wnt、her2/neu、egfr、mapk、erk、mdm2、cdk4、gli1、gli2、igf2、tp53等。示例性tsg包括例如rb、tp53、apc、nf1、brca1、brca2、pten、cdk抑制蛋白(例如pl6、p21)、ptch、wt1等。应当了解的是,这些癌基因和tsg名称中有许多包括多个家族成员并且许多其它tsg是已知的。
在一些实施方案中,疾病是心血管疾病,例如动脉粥样硬化性心脏病或血管疾病、充血性心力衰竭、心肌梗塞、脑血管疾病、外周动脉疾病、心肌病。
在一些实施方案中,疾病是精神疾病、神经疾病或神经发育疾病,例如精神分裂症、抑郁症、双相型情感障碍、癫痫症、自闭症、成瘾。神经退行性疾病包括例如阿尔茨海默氏病、帕金森氏病、肌萎缩性侧索硬化、额颞叶痴呆。[00162]在一些实施方案中,疾病是自身免疫性疾病,例如急性播散性脑脊髓炎、斑秃、抗磷脂综合征、自身免疫性肝炎、自身免疫性心肌炎、自身免疫性胰腺炎、自身免疫性多内分泌腺病综合征、自身免疫性葡萄膜炎、炎症性肠病(克罗恩氏病(crohn'sdisease)、溃疡性结肠炎)、i型糖尿病(例如幼发型糖尿病)、多发性硬化症、硬皮病、强直性脊柱炎、类肉瘤、寻常天疱疮、类天疱疮、银屑病、重症肌无力、系统性红斑狼疮、类风湿性关节炎、幼年型关节炎、银屑病关节炎、白塞氏综合征(behcet'ssyndrome)、莱特尔氏病(reiter'sdisease)、伯格氏病(berger'sdisease)、皮肌炎、多发性肌炎、抗中性粒细胞胞浆抗体相关血管炎(例如伴有多血管炎的肉芽肿病(也被称为韦格纳肉芽肿病(wegener'sgranulomatosis))、显微镜下多血管炎以及丘-施二氏综合征(churg-strausssyndrome))、硬皮病、舍格伦综合征(sjogren'ssyndrome)、抗肾小球基底膜疾病(包括古德帕斯彻氏综合征(goodpasture'ssyndrome))、扩张型心肌病、原发性胆汁性肝硬化、甲状腺炎(例如桥本氏甲状腺炎(hashimoto'sthyroiditis)、格雷夫斯病(graves'disease))、横贯性脊髓炎以及格林-巴利综合征(guillane-barresyndrome)。
在一些实施方案中,疾病是呼吸系统疾病,例如影响呼吸系统的过敏症、哮喘、慢性阻塞性肺病、肺动脉高压、肺纤维化以及类肉瘤病。
在一些实施方案中,疾病是肾脏疾病,例如多囊肾病、狼疮、肾病(肾变病或肾炎)或肾小球肾炎(任何种类)。
在一些实施方案中,疾病是视力丧失或听力丧失,例如与高龄相关。
在一些实施方案中,疾病是感染性疾病,例如由病毒、细菌、真菌或寄生物所引起的任何疾病。
在一些实施方案中,疾病在基因组序列中表现出过度甲基化(例如异常过度甲基化)或非甲基化(例如异常非甲基化)。举例来说,脆性x综合征表现出fmr-1的过度甲基化。dcas9-tet1融合蛋白可以用于特异性地将ccg过度甲基化进行脱甲基化并且重新激活fmg-1,从而治疗脆性x综合征。本文所述的方法可以用于治疗或预防表现出异常甲基化(例如过度甲基化或非甲基化)的疾病或病症。
应当了解的是,本文中的疾病分类不意图具有限制性。本领域的普通技术人员将了解的是,各种疾病可以适当分类为多个不同的组。
在一些实施方案中,疾病是其中已经进行了至少一次全基因组关联(genome-wideassociation,gwa)研究(gwas)的疾病。在一些实施方案中,gwas针对定位于整个基因组或其实质部分(例如基因组的至少80%、90%、95%或更多)中的数千个至数百万个,例如100万个或更多个,例如100万个-500万个或更多个等位基因(例如单核苷酸多态性)对多个“病例”(患有所关注的疾病或其特定表现的受试者)和“对照”(没有所述疾病或表现的受试者)进行分型。应当了解的是,对照数据可以从历史数据中获得。基因分型可以使用微阵列或其它方法进行。由此可以鉴定与疾病(或特定表现)的风险增加(或降低)相关(例如以统计显著性方式)的等位基因。应当了解的是,可以例如使用本领域已知的方法针对多个假设检验来校正统计结果。在一些实施方案中,小于约10"7、10"8或10"9的p值被认为是关联的证据。在一些实施方案中,基因或等位基因或多态性已经在至少一次gwas中被鉴定为促进疾病风险或严重程度。关于与各种疾病相关的gwas研究和遗传变体(等位基因、多态性)的实例,参见例如genome.gov/gwastudies。在一些实施方案中,基因(或任何序列)是其中等位基因或多态性与发生疾病的风险相对于没有所述等位基因或多态性的个体增加或降低到至少1.1倍、1.2倍、1.5倍、2倍、3倍、4倍、5倍、7.5倍、10倍或更多倍有关的基因。在一些实施方案中,等位基因或多态性与发生疾病的风险相对于没有所述等位基因或多态性的个体增加或降低到至少1.1倍、1.2倍、1.5倍、2倍、3倍、4倍、5倍、7.5倍、10倍或更多倍有关。可以促进所关注的任何表型性状,如寿命、体重、对感染的抗性、对各种治疗剂的响应或缺乏响应、对诸如毒素或感染因子(例如病毒、细菌、真菌、寄生物)的潜在有害物质的抗性或易感性的基因、等位基因、多态性或遗传基因座是令人关注的。表型性状可以是体征(如血压);生化标志物,在一些实施方案中,其可以是可在诸如血液、唾液、尿液、泪液等体液中检测的,如代谢物、ldl等的水平,其中标志物的水平异常低或高可能与患有或没有疾病或对疾病的易感性或防止疾病的保护作用相关。
在一些实施方案中,待插入基因组中的序列编码标签。所述序列可以插入基因中适当的位置处以使得产生包含标签的融合蛋白。术语“标签”在广义上使用以包括多种多肽中的任一种。在一些实施方案中,标签包含可用于纯化、表达、增溶和/或检测多肽的序列。在一些实施方案中,标签可以发挥多种功能。在一些实施方案中,标签是相对小的多肽,例如从几个氨基酸到约100个氨基酸长不等。在一些实施方案中,标签具有超过100个氨基酸的长度,例如多达约500个氨基酸的长度或更长。在一些实施方案中,举例来说,标签包括ha、tap、myc、6xhis、flag、v5或gst标签。包含表位(针对其的抗体,例如单克隆抗体是可获得的(例如可商购获得的)或本领域已知的)的标签(例如上述标签中的任一种)可以被称为“表位标签”。在一些实施方案中,标签包括溶解度增强标签(例如sumo标签、nusa标签、snut标签、strep标签或噬菌体t7的ocr蛋白的单体突变体)。参见例如espositod和chatterjeedk.curropinbiotechnol;17(4):353-8(2006)。在一些实施方案中,标签是可切割的,以使得它的至少一部分可以被去除,例如由蛋白酶去除。在一些实施方案中,这是通过在标签中包括蛋白酶切割位点来实现的,所述蛋白酶切割位点例如与所述标签的功能部分相邻或连接。示例性蛋白酶包括例如凝血酶、tev蛋白酶、因子xa、prescission蛋白酶等。在一些实施方案中,使用“自切割”标签。参见例如pct/us05/05763。在一些实施方案中,标签包括荧光多肽(例如gfp或其衍生物,如增强型gfp(egfp))或可以作用于底物以产生可检测信号,例如荧光信号或比色信号的酶。荧光素酶(例如萤火虫荧光素酶、海肾荧光素酶或高斯荧光素酶)是这样的酶的实例。荧光蛋白的实例包括gfp和其衍生物、包含发射不同颜色的光的发色团的蛋白质,如红色荧光蛋白、黄色荧光蛋白以及青色荧光蛋白等。标签,例如荧光蛋白可以是单体的。在某些实施方案中,荧光蛋白是例如sirius、azurite、ebfp2、tagbfp、mturquoise、ecfp、cerulean、tagcfp、mtfpl、mukgl、magl、acgfpl、taggfp2、egfp、mwasabi、emgfp、tagypf、eyfp、topaz、syfp2、venus、citrine、mko、mk02、morange、morange2、tagrfp、tagrfp-t、mstrawberry、mruby、mcherry、mraspberry、mkate2、mplum、nineptune、mtomato、t-sapphire、mametrine、mkeima。关于gfp和许多其它荧光蛋白或发光蛋白的论述,参见例如chalfie,m.和kain,sr(编著)greenfluorescentprotein:properties,applications,andprotocols(methodsofbiochemicalanalysis,第47卷).wiley-interscience,hoboken,n.j.,2006和/或chudakov,dm等,physiolrev.90(3):1103-63,2010。在一些实施方案中,标签可以包含与小分子(例如代谢物)或离子(例如钙、氯离子)结合和/或用作其感受器或细胞内电压、ph值或其它条件的感受器的结构域。可以使用任何可遗传编码的感受器;许多这样的感受器是本领域已知的。在一些实施方案中,可以使用基于fret的感受器。在一些实施方案中,不同的基因被修饰以掺入不同的标签,以使得可区别标记由所述基因编码的蛋白质。举例来说,可以掺入2个至20个不同的标签。在一些实施方案中,所述标签具有不同的发射光谱和/或吸收光谱。在一些实施方案中,标签可以吸收和/或发射红外区或近红外区中的光。应当了解的是,编码标签的任何核酸序列可以经过密码子优化以用于在它待引入的细胞、合子、胚胎或动物中表达。
在一些实施方案中,可能关注的是表达蛋白质的片段或结构域,其可以通过显性失活方式起作用并且可以例如破坏蛋白质的正常功能或相互作用。
在一些实施方案中,所关注的基因编码蛋白质,所述蛋白质的聚集与一种或多种疾病有关,所述疾病可以被称为蛋白质错误折叠疾病。实例包括例如α-突触核蛋白(帕金森氏病和相关病症)、淀粉样蛋白β或τ(阿尔茨海默氏病)、tdp-43(额颞叶痴呆、als)。
在一些实施方案中,所关注的基因编码转录因子、转录共激活因子或共阻遏因子、酶、伴侣蛋白、热激因子、热激蛋白、受体、分泌蛋白、跨膜蛋白、组蛋白(例如hi、h2a、h2b、h3、h4)、外周膜蛋白、可溶性蛋白、核蛋白、线粒体蛋白、生长因子、细胞因子(例如白细胞介素,例如il-1-il-33中的任一种)、干扰素(例如α、β或γ)、趋化因子(例如cxc、cx3c、c(或xc)或cx3c趋化因子)。趋化因子可以是ccl1-ccl28、cxcl1-cxcl17、xcl1或xcl2或cxc3l1)。在一些实施方案中,基因编码集落刺激因子、激素(例如胰岛素、甲状腺激素、生长激素、雌激素、孕酮、睾酮)、细胞外基质蛋白(例如胶原、纤连蛋白)、运动蛋白(例如动力蛋白、肌球蛋白)、细胞粘附分子、主要组织相容性或次要组织相容性(mhc)基因、转运蛋白、通道(例如离子通道)、免疫球蛋白(ig)超家族(igsf)基因(例如编码抗体、t细胞受体、b细胞受体的基因)、肿瘤坏死因子、nf-κb蛋白、整合素、钙粘蛋白超家族成员(例如钙粘蛋白)、选择素、凝血因子、补体因子、纤溶酶原、纤溶酶原激活因子。生长因子包括例如血管内皮生长因子(vegf,例如vegf-a、vegf-b、vegf-c、vegf-d)、表皮生长因子(egf)、胰岛素样生长因子(igf;igf-1、igf-2)、成纤维细胞生长因子(fgf,例如fgf1-fgf22)、血小板衍生生长因子(pdgf)或神经生长因子(ngf)家族的成员。应当了解的是,上述蛋白质家族包括多个成员。任何这样的成员都可以用于各种实施方案中。在一些实施方案中,生长因子促进一种或多种造血细胞类型的增殖和/或分化。举例来说,生长因子可以是csf1(巨噬细胞集落刺激因子)、csf2(粒细胞巨噬细胞集落刺激因子,gm-csf)或csf3(粒细胞集落刺激因子,g-csf)。在一些实施方案中,基因编码促红细胞生成素(epo)。在一些实施方案中,基因编码神经营养因子,即促进神经谱系细胞(如本文所用,该术语包括神经祖细胞、神经元以及神经胶质细胞,例如星形胶质细胞、少突胶质细胞、小胶质细胞)的存活、发育和/或功能的因子。举例来说,在一些实施方案中,所述蛋白质是促进神经突向外生长的因子。在一些实施方案中,所述蛋白质是睫状神经营养因子(cntf)或脑源性神经营养因子(bdnf)。
在一些实施方案中,所关注的基因编码多肽,所述多肽是包含多个亚基的任何蛋白质的亚基。
酶可以是催化已经由国际生物化学与分子生物学联合会(internationalunionofbiochemistryandmolecularbiology)命名委员会(nc-iubmb)指定酶学委员会编号(enzymecommissionnumber)(ec编号)的类型的反应的任何蛋白质。酶包括例如氧化还原酶、转移酶、水解酶、裂解酶、异构酶、连接酶。实例包括例如激酶(蛋白激酶,例如ser/thr激酶、tyr激酶)、脂质激酶(例如磷酸肌醇3-激酶(pi3-激酶或pi3k))、磷酸酶、乙酰转移酶、甲基转移酶、脱乙酰酶、脱甲基酶、脂肪酶、细胞色素p450、葡糖苷酸酶、重组酶(例如rag-1、rag-2)。酶可以参与核苷酸、核酸、氨基酸、蛋白质、神经递质、异生素(例如药物)或其它大分子的生物合成、修饰或降解。
哺乳动物基因组编码至少约500种不同的激酶。激酶可以基于它们的典型底物的性质来分类并且包括蛋白激酶(即将磷酸转移到一种或多种蛋白质的激酶)、脂质激酶(即将磷酸酯基转移到一种或多种脂质的激酶)、核苷酸激酶等。在本发明的某些方面,蛋白激酶(pk)特别受关注。pk基于它们的底物偏好常常被称为丝氨酸/苏氨酸激酶(s/tk)或酪氨酸激酶(tk)。丝氨酸/苏氨酸激酶(ec2.7.11.1)将丝氨酸和/或苏氨酸残基磷酸化,而tk(ec2.7.10.1和ec2.7.10.2)将酪氨酸残基磷酸化。能够将丝氨酸/苏氨酸和酪氨酸残基磷酸化的许多“双重特异性”激酶(ec2.7.12.1)是已知的。人类蛋白激酶家族可以基于序列/结构相似性进一步分为以下组:(1)agc激酶,含有pka、pkc以及pkg;(2)cam激酶,含有钙/调钙蛋白依赖性蛋白激酶;(3)ck1,含有酪蛋白激酶1组;(4)cmgc,含有cdk、mapk、gsk3以及clk激酶;(5)ste,含有酵母sterile7、sterile11以及sterile20激酶的同源物;(6)tk,含有酪氨酸激酶;(7)tkl,含有酪氨酸激酶样激酶组。被称为“非典型蛋白激酶”的另一组含有与其它组缺乏序列同源性,但是已知或被预测具有激酶活性,并且在一些情况下,被预测具有与典型激酶相似的结构折叠的蛋白质。
受体包括例如g蛋白偶联受体、酪氨酸激酶受体、丝氨酸/苏氨酸激酶受体、toll样受体、核受体、免疫细胞表面受体。在一些实施方案中,受体是本文提到的激素、细胞因子、生长因子或分泌蛋白中的任一种的受体。许多g蛋白偶联受体(gpcr)是本领域已知的。参见例如vrolingb,gpcrdb:informationsystemforgprotein-coupledreceptors.nucleicacidsres.2011年1月;39(数据库专刊):d309-19.2010年11月2日电子出版。gpcrdb可以在gpcr.org/7tm/在线找到。g蛋白偶联受体包括例如肾上腺素能受体、大麻素受体、嘌呤能受体、神经肽受体、嗅觉受体。转录因子(tf)(有时被称作序列特异性dna结合因子)与特定的dna序列结合并且(单独或在与其它蛋白质的复合物中),调节转录,例如激活或阻遏转录。示例性tf列于例如
修饰或调节细胞或非人类哺乳动物中的核酸的其它方法描述于pct申请号pct/us2014/034387和美国申请号14/785031中,这些参考文献以引用的方式整体并入本文。
***
本领域技术人员容易了解的是,本发明非常适合于实现所述目的并且获得所提到的结果和优势以及其中固有的那些。本文的描述和实例的细节代表了某些实施方案,是示例性的,并且不意图作为对本发明的范围的限制。其中的修改和其它用途将被本领域技术人员想到。这些修改被包括在本发明的精神内。对于本领域技术人员来说将显而易见的是,可以在不脱离本发明的范围和精神的情况下对本文公开的发明进行不同的替换和修改。
除非明确作相反的说明,否则如本文在说明书和权利要求中所用的冠词“一个/种(a/an)”应当被理解为包括复数指代对象。除非有相反的指示或另外从上下文中显而易见,否则如果一个、多于一个或所有组成员存在于、被用于给定产品或方法中或以其它方式与给定产品或方法相关,那么认为在组的一个或多个成员之间包括“或”的权利要求或描述是满足的。本发明包括其中该组中只有一个成员存在于、被用于给定产品或方法中或以其它方式与给定产品或方法有关的实施方案。本发明还包括其中多于一个或所有的组成员存在于、被用于给定产品或方法中或以其它方式与给定产品或方法有关的实施方案。此外,应当了解的是,除非另外指明或除非对于本领域的普通技术人员来说显然将会出现矛盾或不一致,否则本发明提供了所有变化、组合以及置换,其中来自所列权利要求中的一项或多项的一个或多个限制、要素、条款、描述性术语等被引入依赖于相同基础权利要求(或相关的任何其它权利要求)的另一权利要求中。预期本文所述的所有实施方案在适当的情况下都适用于本发明的所有不同方面。还预期所述实施方案或方面中的任一个在适当时可以自由地与一个或多个其它这样的实施方案或方面组合。在要素以清单形式,例如以马库什组(markushgroup)或类似形式呈现的情况下,应当了解的是,还公开了所述要素的每一个子组,并且任何一个或多个要素可以从该组中被去除。应当了解的是,一般来说,在本发明或本发明的方面被称为包括特定的要素、特征等的情况下,本发明的某些实施方案或本发明的方面由这些要素、特征等组成或基本上由这些要素、特征等组成。为了简单起见,那些实施方案在本文中并未在每种情况下都被一字不差地具体阐述。还应当了解的是,本发明的任何实施方案或方面可以明确地被排除在权利要求之外,无论在说明书中是否叙述了特定的排除。举例来说,可以排除任何一种或多种核酸、多肽、细胞、生物体的物种或类型、病症、受试者或其组合。
在权利要求或描述涉及物质组合物,例如核酸、多肽、细胞或非人类转基因动物的情况下,应当了解的是,除非另外指明或除非对于本领域的普通技术人员来说显然将会出现矛盾或不一致,否则根据本文公开的方法中的任一种制备或使用所述物质组合物的方法以及使用所述物质组合物用于实现本文公开的目的中的任一个的方法是本发明的方面。在权利要求或描述涉及方法的情况下,例如,应当了解的是,除非另外指明或除非对于本领域的普通技术人员来说显然将会出现矛盾或不一致,否则制备可用于执行所述方法的组合物的方法以及根据所述方法制备的产品是本发明的方面。
在本文给出范围的情况下,本发明包括其中包括端点的实施方案、其中不包括两个端点的实施方案以及其中包括一个端点而不包括另一个端点的实施方案。应当假定的是,除非另外指明,否则包括两个端点。此外,应当了解的是,除非另外指明或另外根据上下文和本领域的普通技术人员的理解显而易见,否则被表示为范围的值可以在本发明的不同实施方案中呈现所述范围内的任何具体值或子范围,除非上下文另外明确规定,否则直到所述范围的下限的单位的十分之一为止。还应当了解的是,在本文陈述一系列数值的情况下,本发明包括类似地与该系列中的任何中间值或任何两个值限定的范围相关的实施方案,并且最低的值可以被视为最小值并且最大的值可以被视为最大值。如本文所用的数值包括被表示为百分比的值。对于其中数值以“约”或“大约”开头的本发明的任何实施方案,本发明包括其中叙述精确值的实施方案。对于其中数值没有以“约”或“大约”开头的本发明的任何实施方案,本发明包括其中该值以“约”或“大约”开头的实施方案。除非另有说明或另外从上下文中显而易见,“大约”或“约”一般包括在任一方向上落入与数值相差1%的范围内或在一些实施方案中落入与数值相差5%的范围内或在一些实施方案中落入与数值相差10%的范围内(大于或小于所述数值)的数值(除非这样的数值不允许超过可能值的100%)。应当了解的是,除非明确作相反的说明,否则在本文要求保护的包括多于一个动作的任何方法中,所述方法的动作的顺序不一定限于叙述所述方法的动作的顺序,但是本发明包括其中所述顺序如此受限的实施方案。还应当了解的是,除非另外指明或从上下文中显而易见,否则本文所述的任何产品或组合物可以被认为是“分离的”。
这些方法的具体实例阐述于下文实施例中。
***
实施例:
在本研究中,我们证实了dcas9与tet1酶结构域或dnmt3a的融合分别允许靶向消除或建立dna甲基化。作为原理验证,我们首先诱导了在小鼠胚胎干细胞(mesc)中整合的两个合成甲基化报告基因中的dna甲基化的改变。在应用dcas9-tet1的情况下,我们重新审视了dna甲基化领域中的一些长期存在的问题。我们的结果显示bdnf启动子iv的靶向脱甲基化足以激活它在小鼠皮层神经元中的表达,并且myod远端增强子的靶向脱甲基化促进成纤维细胞重编程为成肌细胞并且促进肌管形成。使用dcas9-dnmt3a,我们证实了ctcf结合位点处的靶向甲基化能够阻断ctcf募集并且改变邻近环中基因的表达,这是通过增加它们与靶向环中绝缘的超级增强子的相互作用频率来实现的。此外,将dcas9-tet1和靶grna向小鼠进行慢病毒递送使得能够通过将启动子脱甲基化来体内激活甲基化报告基因。因此,dcas9-tet1和dcas9-dnmt3a为研究以基因座特异性方式进行dna甲基化的功能意义提供了强有力的工具。
用于编辑dna甲基化的修饰的crispr系统
为了实现dna甲基化的靶向编辑,我们将dcas9与甲基化/脱甲基化途径中的酶融合(图1a)。基于先前使用tale系统靶向特定cpg的研究(bernstein等,2015;maeder等,2013),在我们的系统中选择tet1和dnmt3a作为效应子。序列特异性指导rna(grna)的共表达预期将使dcas9-tet1或dcas9-dnmt3a靶向到特定的基因座并且介导dna甲基化状态的修饰而不改变dna序列。为了优化该嵌合crispr/dcas9-效应子系统,我们测试了在不同位置处具有核定位信号(nls)的两种类型的dcas9-tet1慢病毒构建体:dcas9-nls-tet1和nls-dcas9-nls-tet1(图8a和图8b)。我们还测试了两种类型的grna慢病毒构建体,即被称为grna的广泛使用的嵌合单指导rna(jinek等,2012)和被称为e-grna的具有增强的将cas9引导到所设计的基因组基因座的能力的修饰的指导rna(chen等,2013)。这两种grna构建体都含有由独立cmv启动子驱动的puro选择盒和cherry荧光蛋白盒,这允许在慢病毒转导之后对表达grna的细胞进行荧光激活细胞分选(facs)(图8a)。对这些构建体进行表征显示出dcas9-nls-tet1构建体的稳健的grna诱导的核易位(图8c-图8e),并且因此,选择该构建体用于所有实验以将dna的非特异性修饰减到最低限度。两种类型的grna表现相似(图8c-图8e)并且因此可互换使用。
dcas9-tet1和dcas9-dnmt3a使得能够靶向改变cpg甲基化状态
为了评估dcas9-tet1融合构建体和dcas9-dnmt3a融合构建体是否将分别诱导特定序列的脱甲基化或从头甲基化,我们利用先前在我们的实验室中开发的甲基化报告系统(stelzer等,2015)。该报告系统由控制绿色荧光蛋白(gfp)的表达的合成甲基化感受性启动子(来自印记基因的启动子的保守序列元件,snrpn)组成。将该报告构建体插入基因组基因座中被证实能够如实地报告相邻序列的甲基化状态(stelzer等,2015)。
特定cpg的脱甲基化:为了测试确定的序列是否可以被脱甲基化,我们引入dcas9-tet1构建体与grna的组合以靶向插入dazl启动子中的snrpn-gfp报告基因(图1b和图9a)。dazl是生殖细胞特异性基因,它在es细胞中是过度甲基化的和无活性的,并且因此,gfp报告基因不被表达。为了通过dcas9-tet1激活gfp表达,我们设计了靶向snrpn启动子区域中的所有14个cpg的4个grna。在用共表达dcas9-tet1和4个grna的慢病毒载体感染三天之后,如由从grna构建体表达的cherry阳性信号所标记的一些感染阳性细胞开始开启gfp(图9b)。为了评估dcas9-tet1和靶grna的激活效率,我们使用facs分析了被这两种病毒感染的细胞。在cherry阳性群体中,约26%的具有靶grna的细胞激活了gfp,而只有1%的具有乱序grna的细胞呈gfp阳性(图1c和图9c)。进一步培养这些cherry阳性单细胞以允许形成es细胞集落。具有靶grna的细胞表达gfp,而具有乱序grna的细胞不是这样(图1d)。为了确认这些细胞中gfp的激活是由snrpn启动子的脱甲基化所引起的,我们对来自这些样品的基因组dna进行了亚硫酸氢盐测序。如图1e和图1f中所示,来自具有靶grna的细胞的样品仅在snrpn启动子区域中显示出稳健的脱甲基化,而在相邻的dazl基因座中没有显示出稳健的脱甲基化,并且来自具有乱序grna的细胞的样品显示出与未感染(模拟)对照相似的甲基化状态。我们进一步分析了受感染的cherry阳性细胞内的gfp阳性群体和gfp阴性群体。如图9d中所示,在双阳性细胞(cherry+;gfp+)中观测到snrpn启动子区域的更稳健的脱甲基化。这些结果确认了在增殖细胞中dcas9-tet1和grna靶向消除dna甲基化。
特定cpg的从头甲基化:为了评估dcas9-dnmt3a融合蛋白是否可以将启动子序列从头甲基化并且使基因表达沉默,我们使用在gapdh启动子中携带snrpn-gfp报告基因的细胞。这些细胞呈gfp阳性,这是因为gapdh在es细胞中是非甲基化的并且被表达(stelzer等,2015)。我们将gapdh-snrpn-gfpesc用表达dcas9-dnmt3a和靶向snrpn启动子的grna或乱序grna的慢病毒感染(图2a和图9e),继而进行facs分析。在感染阳性(cherry阳性)群体中,约12%的具有靶grna的细胞使gfp失活,而只有2%的具有乱序grna的细胞呈gfp阴性(图2b和图9f)。当在培养物中培养cherry阳性细胞时,具有靶grna的细胞的gfp表达保持关闭,而具有乱序grna的细胞和模拟对照仍呈gfp阳性(图2c)。此外,亚硫酸氢盐测序显示dcas9-dnmt3a/grna的转导引起snrpn启动子区域中的dna甲基化显著增加,而在相邻的gapdh区域中不是这样(图2d-图2e)。对受感染的cherry阳性细胞内的gfp阳性群体和gfp阴性群体进行进一步分析显示在cherry+;gfp-细胞中snrpn启动子区域的甲基化更稳健(图9g)。为了克服由dcas9-dnmt3a和grna慢病毒的低共转导效率所引起的可能的限制,通过使用piggybac转座子系统将多西环素诱导型dcas9-dnmt3a表达盒整合到gapdh-snrpn-gfpmes细胞系中(图9h)。在递送同一组靶grna之后,facs分析显示gfp失活效率增加到25%(图2f和图9i)。分选的cherry阳性细胞在多西环素处理后显示出gfp表达丧失(图2g)并且在snrpn启动子区域中稳健甲基化(图2h)。我们还产生了新的dcas9-dnmt3a-p2a-bfp构建体,其使得能够通过facs分离表达dcas9-dnmt3a的细胞。在该构建体与grna的慢病毒递送之后,在facs分选的双阳性细胞(bfp+;cherry+)中实现了约70%的gfp失活效率(图9j)。
综上所述,我们的结果表明在分裂细胞中,当通过特异性指导rna靶向时,上述dcas9融合构建体有效地将甲基化的序列脱甲基化(dcas9-tet1)或将非甲基化的序列从头甲基化(dcas9-dnmt3a)。
基于dcas9的甲基化编辑和基于tale的甲基化编辑的比较
为了比较基于dcas9-tet1/dnmt3a的方法与基于tale的方法的甲基化编辑功效和有效范围,我们选择两个先前报道的由基于tale的方法编辑的基因座(bernstein等,2015;maeder等,2013)并且设计使dcas9-tet1/dnmt3靶向到由tale-tet1/dnmt3a结合的相同位点的单个grna。如图10a和图10c中所示,dcas9-dnmt3a和靶向p16基因座的一个单一grna在p16启动子的320bp区域内诱导甲基化平均增加25%,而tale-dnmt3a仅在650bp区域内诱导13%的增加。类似地,dcas9-tet1和靶向rhoxf2基因座的一个单一grna在rhoxf2启动子的150bp区域内诱导甲基化平均减少28%,而tale-tet1仅在200bp区域内诱导14%的减少(图10b-图10c)。这些结果表明dcas9-tet1/dnmt3a系统比基于tale的方法具有更高的甲基化编辑的功效和分辨率。
为了评价dcas9-tet1/dnmt3a介导的甲基化编辑的特异性,我们进行了dcas9chip-seq测定并且在靶向图9a中所述的dazl-snrpn区域的grna存在下鉴定出9个结合位点并且在靶向与mir290基因座相邻的ctcf结合位点(参见下图13a)的grna存在下鉴定出18个结合位点。图10d显示在对于每一组grna所鉴定的结合位点中,靶向的基因座(dazl-snrpn或mir290)显示出对dcas9-dnmt3a的最高水平的结合(表s1)。每一个靶向基因座的第二强的结合位点和第三强的结合位点示于图10e中,并且对这些基因座进行的亚硫酸氢盐测序分析仅显示出甲基化水平的边际变化(图10f-图10g),这可能是因为与靶向的基因座相比,在这些脱靶基因座处dcas9-dnmt3a/tet1的结合亲和力显著更低。这些结果表明基于dcas9的表观遗传编辑可以具有高度特异性。
bdnf启动子iv的靶向脱甲基化激活神经元中的bdnf
已经提出非dna复制依赖性主动脱甲基化在有丝分裂后神经元中起作用(guo等,2011;martinowich等,2003)。为了测试是否可以在有丝分裂后神经元中诱导主动脱甲基化,我们应用dcas9-tet1系统来研究对bdnf基因的调节。bdnf表达可以通过伴有它的启动子iv的脱甲基化的神经元活动来诱导(chen等,2003;martinowich等,2003)。我们设计了靶向bdnf启动子iv中的11个cpg(图11a)的4个grna以确定dcas9-tet1是否可以通过诱导该启动子的脱甲基化来激活bdnf(图3a)。遵循用于产生原代神经元培养物的公认实验程序从e17.5胚胎中分离小鼠皮层神经元并且在体外培养两天(div2)(ebert等,2013)。如图11b-图11d中所示,kcl处理在这些神经元中诱导bdnf表达而没有可检测的细胞增殖。在培养第3天(div3)时将神经元用在存在或不存在4个grna的情况下表达dcas9-tet1的慢病毒载体以差不多100%的转导效率感染(图11e)。在感染后48小时,用kcl处理一些培养物以诱导神经元活动。如图3b-图3c中所示,dcas9-tet1/grna将bdnf表达诱导到约6倍,而在不存在grna的情况下dcas9-tet1仅显示出轻微的诱导作用(小于2倍)并且tet1的无催化活性形式(dc-dt)没有显示出诱导作用。重要的是,同一组dcas9-tet1/grna没有诱导npas4表达(图11f),所述npas4是另一个神经元活动诱导型基因(lin等,2008)。dcas9-tet1与靶向bdnf启动子iv的每一个单独的grna的共转导显示出对bdnf的2倍-3倍诱导(图11g)。我们进行了亚硫酸氢盐测序以研究bdnf启动子iv的甲基化状态。如图3d-图3e中所示,与grna阴性对照相比,dcas9-tet1/grna显著减少该区域中的甲基化,而kcl处理也诱导位置148、66以及19(相对于转录起始位点)处cpg的脱甲基化。
我们的结果证实bdnf启动子iv的脱甲基化可以由dcas9-tet1/grna诱导并且足以激活bdnf表达。由于对于这些实验使用有丝分裂后神经元,因此甲基化的丧失可能是因为主动脱甲基化。为了进一步支持该结论,我们通过tet辅助亚硫酸氢盐测序(tab-seq)分析研究了在dcas9-tet1诱导的脱甲基化的时间过程期间bdnf启动子iv中的5-hmc水平。如图11h中所示,在用dcas9-tet1和grna慢病毒感染后40小时检测到5-hmc并且在60小时之后减少。类似地,在kcl处理之后也检测到5-hmc(图11i)。由于亚硫酸氢盐测序方法不能区分非甲基化的5-胞嘧啶(5-c)和由5-hmc产生的5-甲酰基胞嘧啶/5-羧基胞嘧啶(5-fc/5-cac),因此有可能一些cpg是在用dcas9-tet1/grna靶向之后修饰的5-fc/5-cac。尽管如此,通过用abt-888(parp抑制剂)处理抑制碱基切除修复途径减少了kcl处理对bdnf的激活(图11j),这表明bdnf启动子iv的脱甲基化促进bdnf激活。
为了测试在神经元活动刺激时是否需要内源性tet活性来调节bdnf表达,我们用2-羟基戊二酸盐处理div3神经元,所述2-羟基戊二酸盐是包括tet酶的α-酮戊二酸依赖性双加氧酶的竞争性抑制剂(xu等,2011)。如图11k中所示,tet酶活性的药理学抑制完全消除了kcl处理对bdnf表达的诱导。此外,携带tet1无效突变体的小鼠原代皮层神经元显示出bdnf的激活动力学显著减弱(图11l),这支持了内源性tet对于诱导神经元活动的作用。
myod远端增强子的靶向脱甲基化促进成纤维细胞的成肌重编程
myod作为肌肉发育的主调节因子的作用最初是通过以下观测结果来定义的,即通过5-aza(5-氮杂-2'-脱氧胞苷)处理将成纤维细胞中的dna脱甲基化引起myod的激活以及后续的成肌细胞转化和肌管形成(constantinides等,1977;davis等,1987;lassar等,1986)。已经描述了在myod基因的50kb上游区域内的六个肌肉特异性dmr(schultz等,2015),并且dmr-5与已知的myod远端增强子重叠(brunk等,1996),如图4a中所示。为了测试dmr-5的脱甲基化是否将激活成纤维细胞中的myod,我们设计了靶向该dmr的4个grna(图12a)。在c3h10t1/2细胞中dcas9-tet1与这些grna的共表达引起了对myod表达的中度诱导(3倍),如图4b中所示,所述c3h10t1/2细胞是来自先前用于5-aza介导的myod激活的小鼠胚胎成纤维细胞的亚克隆(constantinides等,1977)。dcas9-tet1/myoddmr-5grna与5-aza处理的组合引起对myod的更高诱导,如图12f中所示。亚硫酸氢盐测序显示用dcas9-tet1和靶grna慢病毒转导的分选的感染阳性细胞的dmr-5区域中的甲基化显著减少,而用无催化活性的tet1(dc-dt)或乱序grna转导的分选的感染阳性细胞不是这样(图4c-图4d)。为了研究myod远端增强子区域的脱甲基化是否可以将成纤维细胞重编程为肌细胞,我们将c3h10t1/2细胞用表达dcas9-tet1和grna的慢病毒感染。将细胞培养14天并且分析myod和mhc(肌球蛋白重链,肌管特异性标志物)表达。如图4e-图4f中所示,dcas9-tet1和靶向dmr-5的grna的共表达诱导中等表达水平的myod,但是不足以在不存在5-aza处理的情况下诱导肌管形成。
我们然后研究了dmr-5的靶向脱甲基化是否将与5-aza处理协同作用以诱导肌管形成(图12b)。为了跟踪在5-aza处理之后肌管形成的过程,进行了时间过程实验。具有异质尺寸的多核肌管(mhc阳性)在处理后14天时开始形成,并且myod阳性细胞比率以及肌管密度和尺寸然后增加直到第25天为止(图12c-图12e)。dcas9-tet1和靶向myoddmr-5的grna的共表达在处理后14天时促进肌管形成,如由与仅表达dcas9-tet1或dc-dt和myoddmr-5grna的细胞相比,显著更成熟的多核mhc+簇(每个mhc+簇>2个核)所证实(图4e、图4g-图4h)。当在处理后的更晚时间点(16天)时分析细胞时进行了类似的观测(图12g-图12j)。我们的结果表明在c3h10t1/2细胞中dcas9-tet1/grna对myod远端增强子的脱甲基化与5-aza协同作用以显著促进成肌细胞转化和肌管形成。
ctcf结合位点的靶向从头甲基化改变ctcf介导的染色质环
ctcf是高度保守的锌指蛋白,其在染色质结构的全局组织中起主要作用(phillips和corces,2009)。转录增强子通常经由形成dna环来与它们的靶基因相互作用(gibcus和dekker,2013;gorkin等,2014;kagey等,2010),这通常被限制在被称作绝缘邻近结构域的更大的ctcf介导的环内(dowen等,2014;ji等,2016;phillips-cremins等,2013),其进而可以形成促成拓扑相关结构域(tad)的环簇(dixon等,2012;nora等,2012)。绝缘邻近结构域的ctcf环锚定位点的缺失可以引起增强子与位于环外部的基因发生不适当的相互作用并且因此增加它们的表达(dowen等,2014)。有趣的是,已经报道ctcf的dna识别位点的甲基化会阻断ctcf结合(bell和felsenfeld,2000;wang等,2012)。为了研究特定ctcf位点的甲基化是否可以改变ctcf介导的染色质环,我们应用dcas9-dnmt3a系统靶向ctcf锚定位点(图5a)。我们设计了使dcas9-dnmt3a靶向到两个ctcf位点的特异性grna(图13)来研究从头甲基化是否将干扰ctcf的成环功能(图5b和图5f)。将多西环素诱导型dcas9-dnmt3ames细胞(图9h)用表达grna的慢病毒感染并且对转导的细胞进行facs分选以用于后续的分析。
使dcas9-dnmt3a靶向到与携带超级增强子的mir290环相邻的ctcf结合位点(图5b)诱导了在该位点处cpg的从头甲基化(图5d-图5e)。对转导细胞进行的基因表达分析显示在该含有超级增强子的绝缘邻近结构域外部并且紧靠靶向的ctcf位点的nlrp12基因显著升高,但是不影响位于mir290环内部的基因或包括au01801和myadm的其它邻近环中的基因的表达(图5c)。类似地,使dcas9-dnmt3a靶向到与携带另一个超级增强子的pou5f1基因环相邻的ctcf结合位点(图5f)诱导了ctcf结合序列中cpg的甲基化(图5h-图5i)并且增加了位于邻近环中并且紧靠靶向的ctcf位点的h2q10的表达,但是不影响pou5f1基因本身或另一个邻近环中的tcf19基因的表达(图5g)。对于任一个靶向的ctcf位点,无催化活性的dnmt3a形式(dc-dd)不像dc-d那样诱导甲基化水平或基因表达的变化(图5c-图5e和图5g-图5i)。这些观测结果与在这些ctcf位点缺失时获得的结果(dowen等,2014)相一致,并且支持了ctcf结合位点的甲基化干扰它的绝缘子功能的观点。
为了测试ctcf结合位点的靶向甲基化是否将引起绝缘的超级增强子与激活基因之间的相互作用频率增加,在这些基因座处进行了染色体构象捕获(3c)测定。如图6a中所示,mir290环中的超级增强子与邻近环中新激活的基因(nlrp12)之间的相互作用频率显著增加,但是nlrp12基因与myadm基因之间的相互作用保持相同,这表明了该靶向ctcf环的开放构象。为了确认增加的相互作用频率归因于阻断ctcf锚定,我们进行了ctcfchip测定。与具有乱序grna、靶向其它ctcf结合位点的grna或无催化活性的dc-dd和mir290靶grna的样品相比,在具有mir290靶grna的样品中ctcf与靶向的基因组位点的结合显著减少(图6b),这支持了dna甲基化阻断ctcf锚定并且因此改变ctcf环构象的观点。对于第二ctcf环(pou5f1环)进行了一组类似的实验,证实了在ctcf结合位点的靶向甲基化之后,绝缘的超级增强子与新激活的基因(h2q10)之间的相互作用频率增加,并且ctcf的结合减少(图6c-图6d)。
综上所述,我们的结果证实dcas9-dnmt3a系统可以用于改变特定ctcf锚定位点的甲基化状态并且因此干扰ctcf成环功能。
dcas9-tet1对内源性基因座进行体内脱甲基化以用于基因激活
为了测试dcas9介导的dna甲基化编辑工具是否可以用于在体内改变甲基化,我们利用先前产生的甲基化敏感性报告小鼠(图7a,参考文献:stelzer等,parent-of-origindnamethylationdynamicsduringmousedevelopment,developmentalcell,在编辑审议中)。在这些转基因小鼠中,将甲基化敏感性snrpn-gfp盒插入dlk1-dio3基因座中以报告它的基因间差异甲基化区域(ig-dmr)的甲基化状态。由于该基因座的ig-dmr在精子发生期间获得父本甲基化,因此gfp报告基因(ig-dmrgf/pat)在携带父本snrpn-gfp等位基因的杂合小鼠中被组成型阻遏(参见参考文献:stelzer等,parent-of-origindnamethylationdynamicsduringmousedevelopment,developmentalcell,在编辑审议中)。如上所示,在mes细胞中dazl基因座中的gfp报告基因通过靶向启动子脱甲基化而被激活(图1)。为了评估在分化的细胞中dlk1-dio3基因座gfp报告基因是否可以由dcas9-tet1激活,我们从ig-dmrgfp/pat转基因小鼠的尾部获得成年小鼠皮肤成纤维细胞,然后将其通过表达dcas9-tet1和snrpn靶grna或乱序grna或tet1的无催化活性形式(dc-dt)和snrpn靶grna的慢病毒转导(图7a)。图7b-图7c中的结果揭示了约80%的cherry(grna)阳性成纤维细胞中的gfp报告基因激活,但是只有在通过dcas9-tet1和snrpngrna慢病毒转导时是这样。对这些细胞进行facs分析进一步确认了这一观点(图14a-图14c)。
为了研究dna甲基化状态是否可以在体内修饰,我们在ig-dmrgfp/pat转基因小鼠的腹侧上用dcas9-tet1和snrpngrna感染了3个表皮部位(图14d)。细胞被稀疏地感染,仅在一些毛囊中看到cherry表达。dcas9-tet1和snrpngrna能够在体内约85%受感染的皮肤真皮细胞中激活gfp报告基因表达,而dcas9-tet1和乱序grna或dc-dt和snrpngrna不能(图7h、图14e-图14f)。此外,我们使用立体定位装置将ig-dmrgfp/pat转基因小鼠的脑用慢病毒载体感染并且通过共聚焦显微镜术分析对脑切片中的靶向dna甲基化的影响。为了消除可能的个体间变异性,我们将表达dcas9-tet1和snrpngrna的慢病毒载体以及两种阴性对照载体组合注射到同一脑的不同区域中(图7d)。如图7e-图7f中所示,在如由cherry表达所示,用所有三种慢病毒组合感染之后,只有表达dcas9-tet1和snrpngrna的慢病毒载体以约70%的激活效率激活gfp报告基因,而表达dcas9-tet1和scgrna或dc-dt和snrpngrna的载体不是这样(图7g)。
讨论
在本研究中,我们已经重新利用crispr/cas9系统来编辑基因组序列的甲基化状态。使无催化活性的cas9蛋白(dcas9)与tet1催化结构域(dcas9-tet1)或dnmt3a(dcas9-dnmt3a)融合以可预测地改变靶序列的表观遗传状态。当被dcas9-tet1靶向时,插入甲基化和沉默的dazl基因的启动子区域中的gfp报告基因被脱甲基化并且被激活,而当被dcas9-dnmt3a靶向时,插入活性和非甲基化的gapdh基因的启动子区域中的gfp报告基因被从头甲基化并且沉默。当使dcas9-tet1靶向到有丝分裂后神经元中的无活性bdnf启动子iv时,所述启动子变成脱甲基化的并且被激活。重要的是,该工具可预测地改变调节区域的甲基化状态和活性:无活性的myod远端增强子的靶向脱甲基化激活了所述基因并且促进肌肉分化,并且ctcf锚定位点的靶向甲基化抑制ctcf结合并且干扰它作为染色质环之间的绝缘子的功能。最终,所述编辑工具可以在体内改变调节序列的甲基化状态,这是因为将dcas9-tet1和靶grna的慢病毒载体注射到转基因小鼠的真皮或脑中将dlk1-dio3印记基因座中甲基化的snrpn启动子脱甲基化并且激活甲基化感受性gfp报告基因。
已经提出动态dna甲基化来解码神经元活动(sweatt,2013)。举例来说,用kcl处理神经元已经被证实会将bdnf启动子iv去沉默并且诱导与启动子区域中一些甲基化的cpg的脱甲基化相关的bdnf表达(chen等,2003;martinowich等,2003)。当bdnf启动子iv被dcas9-tet1靶向时,观测到甲基化的cpg的广泛脱甲基化,并且bdnf被激活到与在用kcl处理培养物时相似的水平。由于所述神经元是有丝分裂后的,因此dcas9-tet1介导的启动子序列脱甲基化可能是主动脱甲基化的结果,如先前所提出的那样(wu和zhang,2014)。尽管有可能bdnf启动子中的一些cpg是在用dcas9-tet1/grna靶向之后修饰的5-fc/5-cac,但是通过抑制ber途径阻断5-fc/5-cac恢复成非甲基化的胞嘧啶减少了bdnf表达,这表明bdnf启动子iv的脱甲基化促进bdnf的激活。重要的是,我们的结果确定了bdnf启动子iv的脱甲基化与基因激活之间的因果关系。
dna甲基化作为细胞谱系之间的屏障的作用与先前的观测结果相一致,即通过用5-aza处理将成纤维细胞中的dna脱甲基化可以激活myod并且介导肌管形成(constantinides等,1977;davis等,1987;lassar等,1986)。使dcas9-tet1靶向到成纤维细胞中甲基化的myod远端增强子诱导了cpg的脱甲基化并且引起myod的中度激活,但是未能产生成肌细胞。然而,当将dcas9-tet1/grna慢病毒转导与5-aza处理组合时,与单独的5-aza处理相比,观测到显著增强的成肌细胞和肌管形成。已经鉴定出myod上游另外的dmr(schultz等,2015)并且有可能的是,可能需要这些位点与远端增强子的组合的脱甲基化来诱导成纤维细胞有效转化成成肌细胞。
对哺乳动物染色体结构的最新研究揭示了染色质被组织成由诸如粘连蛋白和ctcf的染色质结构蛋白介导的拓扑相关结构域和基因环(ji等,2016;seitan等,2013;sofueva等,2013;tang等,2015;zuin等,2014)。新出现的数据表明高阶染色质结构在发育期间赋予表观遗传信息并且在癌症中经常发生改变(flavahan等,2016;ji等,2016;narendra等,2015)。据报道,当ctcf的识别序列被甲基化时,ctcf的结合受到抑制(bell和felsenfeld,2000;kang等,2015;wang等,2012)。使dcas9-dnmt3a靶向到两个ctcf结合位点诱导了这些位点处cpg的从头甲基化并且干扰蛋白质的绝缘子功能,如由靶向环中绝缘的超级增强子与邻近环中的基因之间的相互作用频率增加,从而引起这些基因上调所证实。这表明dcas9-dnmt3a系统是操纵染色质结构并且评估它在发育期间和在疾病背景下的功能意义的有用的工具。
我们的结果表明与表观遗传效应子tet1和dnmt3a融合的dcas9代表了用于编辑特定基因组序列的dna甲基化的强有力的工具箱。这些工具与基于tale的方法的比较显示出更高的甲基化编辑的功效和分辨率,并且进行dcas9chip-seq,之后对潜在脱靶结合基因座进行亚硫酸氢盐测序揭示了甲基化水平的边际变化,这表明了可以使用适当设计的grna实现高度特异性。这些dcas9-dnmt3a/tet1工具将可用于深入了解dna甲基化在诸如基因表达、细胞命运决定以及高阶染色质结构的组织的不同生物过程中的功能意义。
实验程序
质粒设计和构建
将pcr扩增的来自pjfa344c7(addgene质粒:49236)的tet1催化结构域、来自mlm3739(addgene质粒:49959)的tet1无活性催化结构域或来自pcdna3-hdnmt3a(addgene质粒:35521)的dnmt3a克隆到具有bamhi位点和ecori位点的修饰的pdcas9质粒(addgene质粒:44246)中。然后,对dcas9-nls-tet1或dcas9-nls-dnmt3a进行pcr扩增并且克隆到具有asci和ecori的fuw载体(addgene质粒:14882)中以包装慢病毒。通过将退火的寡核苷酸(nls)插入到具有xbai和asci的fuw-dcas9-nls-tet1中来克隆nls-dcas9-nls-tet1。通过将退火的寡核苷酸插入到具有aari位点的修饰的pgrna质粒(addgene质粒:44248)中来克隆grna表达质粒。通过使pcr扩增的来自fuw构建体的dcas9-nls-tet1或dcas9-nls-dnmt3a与具有nhei和ecori的修饰的piggybac转座子载体(wilson等,2007)连接来克隆piaggybac-dcas9-tet1和piaggybac-dcas9-dnmt3a。在转染之前对所有构建体进行测序。关于grna设计和构建的引物信息列于补充表s2中。相关质粒已经被存入addgene质粒数据库中。靶向p16基因座的tale-dnmt3a构建体是由klauskaestner博士馈赠并且靶向rhoxf2基因座的tale-tet1来自addgene公司(质粒编号:49943)。dcas9-dnmt3a和dcas9-tet1cd以及它们的突变体的全长蛋白质序列列于补充表s6中。
细胞培养、慢病毒产生以及稳定细胞系产生
使用标准esc培养基将小鼠胚胎干细胞(mesc)培养在经过辐照的小鼠胚胎成纤维细胞(mef)上:(500ml)dmem,补充有10%fbs(hyclone公司)、10ug重组白血病抑制因子(lif)、0.1mmβ-巯基乙醇(sigma-aldrich公司)、青霉素/链霉素、1mml-谷氨酰胺以及1%非必需氨基酸(全部都来自invitrogen公司)。将c3h10t1/2细胞培养在含有10%fbs的标准deme培养基中。通过将hek293t细胞用fuw构建体或pgrna构建体以及标准包装载体(pcmv-dr8.74和pcmv-vsvg)转染,继而进行基于超速离心的浓缩来产生表达dcas9-tet1、dcas9-dnmt3a以及grna的慢病毒。病毒滴度(t)是基于293t细胞的感染效率计算的,其中t=(p*n)/(v),t=滴度(tu/ul),p=根据荧光标记的感染阳性细胞%,n=转导时的细胞数,v=使用的病毒总体积。请注意,tu表示转导单位。为了产生具有整合的多西环素诱导型dcas9-tet1或dcas9-dnmt3a转基因的稳定细胞系,根据供应商的方案,将piggybac-dcas9-tet1或piggybac-dcas9-dnmt3a构建体与表达转座酶的辅助质粒一起使用x-tremegene9转染试剂(roche公司)转染到c3h10t1/2细胞中或使用xfect转染试剂(clontech公司)转染到mesc细胞中。使用g418(400ug/ml)选择稳定整合的细胞10天。成年小鼠成纤维细胞源自于ig-dmrgfp/pat报告小鼠的尾部。简单地说,从携带父本传递的ig-dmr-snrpn-gfp甲基化报告基因的3个月大的小鼠获得约2cm长的小鼠尾部,并且通过70%etoh灭菌。在37℃在15ml锥形离心管中将约2mm×2mm切碎的尾部块用5ml的1mg/ml胶原酶iv消化90分钟。将5mlmef培养基添加到管中以终止消化。使用注射器塞在平缓研磨下将解离的细胞通过40um细胞过滤器挤出。然后收集细胞并且培养用于病毒感染。在本研究中感染后3天时分析细胞。
小鼠品系和育种策略
tet1突变型小鼠先前在我们的实验室中产生(dawlaty等,2011)。所述研究中的tet1ko小鼠维持在混合129和c57bl/6背景下。为了获得tet1ko小鼠,使对于tet1杂合的雄性小鼠和雌性小鼠杂交。为了获得野生型小鼠原代皮层神经元,使雄性c57bl/6小鼠和雌性c57bl/6小鼠交配。如所述产生ig-dmrgfp/pat甲基化报告小鼠品系(参考文献:stelzer等,parent-of-origindnamethylationdynamicsduringmousedevelopment,developmentalcell,在编辑审议中)。使具有ig-dmrgfp/pat报告基因等位基因的雄性小鼠与c57bl/6雌性杂交以产生携带父本传递的等位基因的成年后代用于体内dna甲基化编辑分析。根据机构指南处理小鼠并且由麻省理工学院(massachusettsinstituteoftechnology)的动物护理委员会(committeeonanimalcare,cac)和比较医学系(departmentofcomparativemedicine,dcm)批准。
小鼠的病毒感染和组织样品制备
根据机构指南用适当的慢病毒混合物感染小鼠并且由麻省理工学院的动物护理委员会(cac)和比较医学系(dcm)批准。确切地说,为了感染小鼠皮肤,通过hamilton注射器将表达dcas9-tet1和scgrna、dc-dt的无活性突变体和靶grna以及dcas9-tet1和靶grna的慢病毒递送到深度麻醉的携带父本ig-dmrgfp报告基因等位基因的小鼠的腹侧上的多个真皮部位中(图14d)。为了感染小鼠脑,通过立体定位装置(leicabiosystems公司,benchmark具有手动精细驱动器的数字立体定位仪)将各种慢病毒混合物递送到深度麻醉的携带父本ig-dmrgfp/pat报告基因等位基因的小鼠的以下位置中(相对于franklin和paxinos小鼠脑图谱)(图7d):dcas9-tet1和scgrna(a-p0.70mm,m-l1.50mm,d-v1.50mm);dc-dt的无活性突变体和snrpngrna(a-p-1.90mm,m-l-1.50mm,d-v1.50mm);以及dcas9-tet1和snrpngrna(a-p-1.90mm,m-l1.50mm,d-v1.50mm)。dc-t/dc-dt和grna慢病毒的滴度分别是1.2×104tu/ul和1.2×105tu/ul。在感染后3天时将小鼠处死。通过穿心灌注4%多聚甲醛(pfa)/pbs来固定动物。解剖固定的皮肤垫和脑样品并且在4℃用4%多聚甲醛(pfa)/pbs后固定过夜。用振动切片机(leicavt1100)以150um厚度对脑样品进行切片并且用恒冷切片机(leica公司)以10um厚度对皮肤样品进行切片,继而进行免疫组织化学分析。对于振动切片机切片,将组织包埋在3%琼脂糖凝胶中。对于冷冻切片,将组织在30%蔗糖/pbs中平衡,之后包埋在最佳切割温度(optimalcuttingtemperature,oct)配混物中。
免疫组织化学、显微镜术以及图像分析
在室温下将神经元、hek293t细胞、小鼠es细胞以及c3h10t1/2细胞用4%多聚甲醛(pfa)固定10分钟。将细胞用pbst(含有0.1%tritonx-100的1×pbs溶液)透化,之后用于pbst中10%的正常驴血清(nds)封闭。然后将细胞与适当稀释的一抗在含有5%nds的pbst中在室温下孵育1小时或在4℃孵育12小时,在室温下用pbst洗涤3次,然后与所期望的二抗在含有5%nds和dapi的tbst中孵育以复染核。将细胞用pbst洗涤3次,之后用fluoromountg(southernbiotech公司)封埋到载片上。先前描述了用于组织切片的免疫染色程序(wu等,2014a)。简单地说,在室温下将切片用pbst(含有0.5%tritonx-100的1×pbs溶液)透化1小时,之后用于pbst中10%的正常驴血清(nds)封闭。然后在4℃将切片与所期望的一抗在含有5%nds的pbst中孵育24小时,在室温下用pbst洗涤3次,然后与二抗在含有5%nds和dapi的tbst中孵育以复染核。将切片用pbst洗涤3次,之后进行载片封埋。在本研究中使用以下抗体:鸡抗gfp(1:1000,aveslabs公司)、小鼠抗cas9(7a9,1:1000,emdmillipore公司)、兔抗bdnf(1:1000,thermofisher公司)、鸡抗map2(1:1000,sigmaaldrich公司)、小鼠抗tuj1(1:1000,r&dsystem公司)、兔抗myod(c-20,1:1000,santacruzbiotechnology公司)、小鼠抗mhc(mf20,1:1000,fisherscientific公司)、小鼠抗myog(f5g,1:1000,lifetechnologies公司)。在zeisslsm710共聚焦显微镜上拍摄图像并且使用zen软件、imagej/fiji以及adobephotoshop处理。对于基于成像的定量,除非另外说明,否则对3-5个代表性图像进行定量并且使用excel或graphpad将数据绘制成平均值±sd。
facs分析
为了评估在处理之后gfp和/或cherry阳性细胞的比例,将经过处理的细胞用胰蛋白酶解离并且在生长培养基中制备单细胞悬浮液,在怀特黑德研究所流式细胞术核心(whiteheadinstituteflowcytometrycore)根据制造商的方案经受bdfacsaria细胞分选仪。使用flowjo软件分析数据。
小鼠原代皮层神经元培养、edu标记以及神经诱导
如先前所述从野生型或tet1ko小鼠胚胎产生解离的e17.5皮层神经元培养物(ebert等,2013)。简单地说,在含有1×青霉素/链霉素(gibco公司:15140122)、1×丙酮酸盐(gibco公司:11360070)以及30mm葡萄糖的冰冷的1×hbss(gibco14185-052)中解剖e17.5皮层。将组织切碎到约1mm3并且用木瓜蛋白酶神经组织解离系统(worthingtonbiochemicals公司)遵循制造商的说明书解离。将细胞重悬在nm5培养基(5%fbs(hyclone公司)、2%b27补充剂(gibco17504044)、1×青霉素/链霉素以及1×glutamaxi(gibco35050-061))中。将1×106个细胞平板接种到包被有聚d-赖氨酸(pdl,sigma公司)的6孔板的每个孔中。在div2时,用2.5umarac(sigmac-6645)将细胞处理过夜以消除有丝分裂星形胶质细胞和神经祖细胞的过度细胞分裂。在div3时向培养物供给新鲜nm5培养基,随后用50mmkcl进行膜去极化或用优选的慢病毒感染。我们在体外培养的最开始时开始处理,因此ap5和ttx(河豚毒素)处理的步骤使培养物中的基础活动沉默,之后省略了kcl处理。对于edu标记,将原代神经元培养物用10um的最终浓度的edu处理24小时,继而根据制造商的说明书(thermofisherscientific公司)进行click-itedu标记程序。将细胞固定用于免疫组织化学分析,在trizol中裂解以提取总rna用于rt-qpcr或裂解以提取dna用于亚硫酸氢盐测序分析。
成纤维细胞向成肌细胞转化测定
先前描述了成肌细胞转化测定(constantinides等,1977)。简单地说,将c3h10t1/2小鼠胚胎成纤维细胞以每孔1×104个细胞平板接种到6孔板中,然后用表达dcas9-tet1和靶grna的慢病毒感染。在感染后24小时,将细胞用媒介物对照(hepes缓冲液)或5-氮杂胞苷(1um)处理24小时,并且在不同的时间点收获用于随后分析。基于人类/小鼠基因组同源性定义小鼠myod基因上游的dmr(schultz等,2015)。
蛋白质印迹法
通过x-tremegene9试剂,遵循制造商的方案将hek293t细胞用各种构建体转染。在转染后2天时,用蛋白酶抑制剂(invitrogen公司)通过ripa缓冲液裂解细胞,并且进行标准免疫印迹分析。使用小鼠抗cas9抗体(1:1000,activemotif公司)和小鼠α-微管蛋白抗体(1:1000,sigma公司)。
rt-qpcr
根据制造商的说明书,使用trizol,继而使用direct-zol(zymoresearch公司)收获细胞。使用第一链cdna合成(invitrogensuperscriptiii)将rna转化成cdna。使用sybrgreen(invitrogen公司)准备定量pcr反应并且在7900htfastabi仪器中进行。关于rt-qpcr的引物信息列于补充表s3中。
chip测定
如先前所述进行chip实验(dowen等,2014)。简单地说,在室温下通过于培养基中1%的甲醛使细胞交联10分钟,然后通过添加0.125m甘氨酸淬灭5分钟。将收集的细胞用pbs洗涤两次,然后重悬在3.5ml超声处理缓冲液中。在冰水混合物中以0.5分钟脉冲启动和1分钟停止以及24瓦进行超声处理10个循环。然后,在4℃以14,000×rpm将细胞裂解物旋降10分钟。保存50ul的上清液作为gdna的输入。添加10ul的抗ctcf抗体(emdmillipore公司:07729)或抗cas9抗体(activemotif公司)并且在4℃孵育过夜。将50ul蛋白g磁珠(dynabead)添加到抗体-细胞裂解物混合物中并且在4℃孵育过夜。然后,将珠粒用超声处理缓冲液、含有高盐(500mmnacl)的超声处理缓冲液、licl洗涤缓冲液以及te缓冲液洗涤。通过在65℃烘箱中孵育15分钟从珠粒上洗脱结合的蛋白质-dna复合物,然后在65℃下反向交联过夜。使用qiagenqiaquickpcr纯化试剂盒纯化结合的dna,然后进行qpcr分析或测序。
chip-seq数据分析
使用先前报道的方法分析测序数据(wu等,2014b)。将读段解复用并且使用star(dobin等,2013)将前25个碱基定位到小鼠基因组(mm10),其需要允许一个错配的唯一定位。将定位的读段折叠并且从每一个样品中随机采样相同数量的读段(约1500万个)以匹配测序深度。使用macs(zhang等,2008)以默认设置呼叫峰。对于每一个样品,其它五个样品各自用作对照并且只有在所有五个对照中呼叫的峰被定义为候选峰。通过相对于背景的富集倍数来过滤候选峰并且选择阈值以使得在四个对照样品(输入、模拟ip、单独的dcas9以及乱序grna)中没有峰通过该阈值。请注意,输入中被定位到45srrna和线粒体dna的六个候选峰被排除在分析之外。原始数据可在以下链接中获得:ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ktohskmgnhudhud&acc=gse83890。
亚硫酸氢盐转化、pcr以及测序
使用epitect亚硫酸氢盐试剂盒(qiagen公司),遵循制造商的说明书建立dna的亚硫酸氢盐转化。通过第一轮巢式pcr扩增所得的修饰的dna,之后使用基因座特异性pcr引物进行第二轮(补充表s3)。第一轮巢式pcr如下进行:94℃持续4分钟;55℃持续2分钟;72℃持续2分钟;将步骤1-3重复1次;94℃持续1分钟;55℃持续2分钟;72℃持续2分钟;将步骤5-7重复35次;72℃持续5分钟;保持12℃。第二轮pcr如下:95℃持续4分钟;94℃持续1分钟;55℃持续2分钟;72℃持续2分钟;将步骤2-4重复35次;72℃持续5分钟;保持12℃。将所得的扩增产物进行凝胶纯化,亚克隆到pcr2.1-topo-ta克隆载体(lifetechnologies公司)中,并且测序。关于亚硫酸氢盐测序的引物信息列于补充表s4中。
基因座特异性tab-seq
如先前所述进行tab-seq(yu等,2012)。简单地说,在37℃将1ug来自经过处理的小鼠皮层神经元的基因组dna在含有50mmhepes缓冲液(ph8.0)、25mmmgcl2、100ng/ml模型dna、200mmudp-glc以及1mmbgt的溶液中葡糖基化1小时。在反应之后,将dna进行柱纯化。在含有50mmhepes缓冲液(ph8.0)、100mm硫酸铵铁(ii)、1mma-酮戊二酸盐、2mm抗坏血酸、2.5mmdtt、100mmnacl、1.2mmatp、15ng/ml葡糖基化的dna以及3mm重组mtet1的溶液中进行氧化反应。将反应物在37℃孵育1小时。在蛋白酶k处理之后,将dna进行柱纯化,然后遵循供应商的说明书,将其应用于epitect亚硫酸氢盐试剂盒(qiagen公司)。通过第一轮巢式pcr扩增所得的修饰的dna,之后使用基因座特异性pcr引物进行第二轮(补充表s3)。将所得的扩增产物进行凝胶纯化,亚克隆到pjet克隆载体(lifetechnologies公司)中并且测序。关于亚硫酸氢盐测序的引物信息列于补充表s4中。
染色体构象捕获(3c)测定
在室温下将5×106个mesc用1%甲醛固定20分钟,并且在室温下通过0.125m甘氨酸将反应淬灭5分钟。收集交联的细胞并且用1ml冰冷pbs洗涤。将细胞沉淀物用550μl裂解缓冲液(10mmtris-hcl(ph8.0)、10mmnacl以及0.2%igepalca630和蛋白酶抑制剂)重悬,并且在冰上孵育20分钟。然后将细胞沉淀物用1×neb缓冲液2(neb公司,b7002s)洗涤两次,然后在62℃与50μl的0.5%sds一起孵育10分钟。在加热后,将145μl的h2o和25μl的10%tritonx-100添加到混合物中并且在37℃孵育15分钟。添加25μl的10×neb缓冲液2和100u的bglii(neb公司,r0144s)以在37℃消化染色质过夜。通过在62℃孵育20分钟来使消化反应失活。然后,添加713μl的h2o、120μl的10×t4dna连接酶缓冲液(neb公司,b0202)、100μl的10%tritonx-100、12μl的10mg/mlbsa以及5μl的t4dna连接酶(neb公司,m0202)并且在16℃孵育22小时。将染色质反向交联,并且通过苯酚:氯仿:异戊醇(sigma公司,p3803)萃取来纯化dna。通过定量实时pcr,使用定制的taqman探针分析mir290基因座和pou5f1基因座处的3c相互作用(图6a和图6c)。将qpcr反应中dna的量在3c文库中使用针对actb基因座的定制taqman探针标准化。引物和探针序列列于补充表s5中。
补充表:
补充表s1:dcas9chip-seq(seqidno:1-35)
表图例说明
rhr:染色体
起点:峰的起点坐标
终点:峰的终点坐标
指导序列匹配数:与指导序列的碱基匹配数
指导序列匹配+ngg:峰内的指导序列匹配序列+ngg(错配序列以小写字母表示)
峰得分:macs峰得分(对数转换:-10×log10(p值))
峰顶高度:macs峰顶高度
相对结合:相对于靶grna的峰顶高度标准化
补充表s2:用于构建指导rna的引物序列
补充表s3:qpcr引物
补充表s4:亚硫酸氢盐测序引物
补充表s5:3c测定引物
补充表s6:dcas9-dnmt3a/tet和突变体的全长蛋白质序列
dcas9-dnmt3a_im(dc-dd,dnmt3a的无活性突变形式)(seqidno:158)
dcas9-tet1cd_im(dc-dt,tet1的无活性突变形式)(seqidno:160)
参考文献(其内容在此以引用的方式整体并入本文):
bell,a.c.,andfelsenfeld,g.(2000).methylationofactcf-dependentboundarycontrolsimprintedexpressionoftheigf2gene.nature405,482-485.
bernstein,d.l.,lelay,j.e.,ruano,e.g.,andkaestner,k.h.(2015).tale-mediatedepigeneticsuppressionofcdkn2aincreasesreplicationinhumanfibroblasts.jclininvest125,1998-2006.
bird,a.(2002).dnamethylationpatternsandepigeneticmemory.genesdev16,6-21.
boch,j.,andbonas,u.(2010).xanthomonasavrbs3family-typeiiieffectors:discoveryandfunction.annualreviewofphytopathology48,419-436.
brunk,b.p.,goldhamer,d.j.,andemerson,c.p.,jr.(1996).regulateddemethylationofthemyoddistalenhancerduringskeletalmyogenesis.devbiol177,490-503.
carroll,d.(2008).progressandprospects:zinc-fingernucleasesasgenetherapyagents.genether15,1463-1468.
cedar,h.,andbergman,y.(2012).programmingofdnamethylationpatterns.annurevbiochem81,97-117.
chen,b.,gilbert,l.a.,cimini,b.a.,schnitzbauer,j.,zhang,w.,li,g.w.,park,j.,blackbum,e.h.,weissman,j.s.,qi,l.s.,etal.(2013).dynamicimagingofgenomiclociinlivinghumancellsbyanoptimizedcrispr/cassystem.cell155,1479-1491.
chen,w.g.,chang,q.,lin,y.,meissner,a.,west,a.e.,griffith,e.c.,jaenisch,r.,andgreenberg,m.e.(2003).derepressionofbdnftranscriptioninvolvescalcium-dependentphosphorylationofmecp2.science302,885-889.
choudhury,s.r.,cui,y.,lubecka,k.,stefanska,b.,andirudayaraj,j.(2016).crispr-dcas9mediatedtet1targetingforselectivednademethylationatbrca1promoter.oncotarget.
cong,l.,ran,f.a.,cox,d.,lin,s.l.,barretto,r.,habib,n.,hsu,p.d.,wu,x.b.,jiang,w.y.,marraffini,l.a.,etal.(2013).multiplexgenomeengineeringusingcrispr/cassystems.science339,819-823.
constantinides,p.g.,jones,p.a.,andgevers,w.(1977).functionalstriatedmusclecellsfromnon-myoblastprecursorsfollowing5-azacytidinetreatment.nature267,364-366.
davis,r.l.,weintraub,h.,andlassar,a.b.(1987).expressionofasingletransfectedcdnaconvertsfibroblaststomyoblasts.cell51,987-1000.
dawlaty,m.m.,breiling,a.,le,t.,barrasa,m.i.,raddatz,g.,gao,q.,powell,b.e.,cheng,a.w.,faull,k.f.,lyko,f.,etal.(2014).lossoftetenzymescompromisesproperdifferentiationofembryonicstemcells.devcell29,102-111.
dawlaty,m.m.,ganz,k.,powell,b.e.,hu,y.c.,markoulaki,s.,cheng,a.w.,gao,q.,kim,j.,choi,s.w.,page,d.c.,etal.(2011).tetlisdispensableformaintainingpluripotencyanditslossiscompatiblewithembryonicandpostnataldevelopment.cellstemcell9,166-175.
dejager,p.l.,srivastava,g.,lunnon,k.,burgess,j.,schalkwyk,l.c.,yu,l.,eaton,m.l.,keenan,b.t.,ernst,j.,mccabe,c.,etal.(2014).alzheimer′sdisease:earlyalterationsinbraindnamethylationatank1,bin1,rhbdf2andotherloci.natneurosci17,1156-1163.
dixon,j.r.,selvaraj,s.,yue,f.,kim,a.,li,y.,shen,y.,hu,m.,liu,j.s.,andren,b.(2012).topologicaldomainsinmammaliangenomesidentifiedbyanalysisofchromatininteractions.nature485,376-380.
dobin,a.,davis,c.a.,schlesinger,f.,drenkow,j.,zaleski,c.,jha,s.,batut,p.,chaisson,m.,andgingeras,t.r.(2013).star:ultrafastuniversalrna-seqaligner.bioinformatics29,15-21.
doi,a.,park,i.h.,wen,b.,murakami,p.,aryee,m.j.,irizarry,r.,herb,b.,ladd-acosta,c.,rho,j.,loewer,s.,etal.(2009).differentialmethylationoftissue-andcancer-specificcpgislandshoresdistinguisheshumaninducedpluripotentstemcells,embryonicstemcellsandfibroblasts.natgenet41,1350-1353.
dowen,j.m.,fan,z.p.,hnisz,d.,ren,g.,abraham,b.j.,zhang,l.n.,weintraub,a.s.,schuijers,j.,lee,t.i.,zhao,k.,etal.(2014).controlofcellidentitygenesoccursininsulatedneighborhoodsinmammalianchromosomes.cell159,374-387.
ebert,d.h.,gabel,h.w.,robinson,n.d.,kastan,n.r.,hu,l.s.,cohen,s.,navarro,a.j.,lyst,m.j.,ekiert,r.,bird,a.p.,etal.(2013).activity-dependentphosphorylationofmecp2threonine308regulatesinteractionwithncor.nature499,341-345.
flavahan,w.a.,drier,y.,liau,b.b.,gillespie,s.m.,venteicher,a.s.,stemmer-rachamimov,a.o.,suva,m.l.,andbernstein,b.e.(2016).insulatordysfunctionandoncogeneactivationinidhmutantgliomas.nature529,110-114.
gibcus,j.h.,anddekker,j.(2013).thehierarchyofthe3dgenome.molcell49,773-782.gilbert,l.a.,larson,m.h.,morsut,l.,liu,z.,brar,g.a.,torres,s.e.,stern-ginossar,n.,brandman,o.,whitehead,e.h.,doudna,j.a.,etal.(2013).crispr-mediatedmodularrna-guidedregulationoftranscriptionineukaryotes.cell154,442-451.
gorkin,d.u.,leung,d.,andren,b.(2014).the3dgenomeintranscriptionalregulationandpluripotency.cellstemcell14,762-775.
guo,j.u.,su,y.,zhong,c.,ming,g.l.,andsong,h.(2011).hydroxylationof5-methylcytosinebytet1promotesactivednademethylationintheadultbrain.cell145,423-434.
hilton,i.b.,d′ippolito,a.m.,vockley,c.m.,thakore,p.i.,crawford,g.e.,reddy,t.e.,andgersbach,c.a(2015).epigenomeeditingbyacrispr-cas9-basedacetyltransferaseactivatesgenesfrompromotersandenhancers.natbiotechnol33,510-517.
hockemeyer,d.,soldner,f.,beard,c.,gao,q.,mitalipova,m.,dekelver,r.c.,katibah,g.e.,amora,r.,boydston,e.a.,zeitler,b.,etal.(2009).efficienttargetingofexpressedandsilentgenesinhumanescsandipscsusingzinc-fingernucleases.natbiotechnol27,851-857.
hockemeyer,d.,wang,h.,kiani,s.,lai,c.s.,gao,q.,cassady,j.p.,cost,g.j.,zhang,l.,santiago,y.,miller,j.c.,etal.(2011).geneticengineeringofhumanpluripotentcellsusingtalenucleases.natbiotechnol29,731-734.
jaenisch,r.,andbird,a.(2003).epigeneticregulationofgeneexpression:howthegenomeintegratesintrinsicandenvironmentalsignals.natgenet33suppl,245-254.
ji,x.,dadon,d.b.,powell,b.e.,fan,z.p.,borges-rivera,d.,shachar,s.,weintraub,a.s.,hnisz,d.,pegoraro,g.,lee,t.i.,etal.(2016).3dchromosomeregulatorylandscapeofhumanplufipotentcells.cellstemcell18,262-275.
jinek,m.,chylinski,k.,fonfara,i.,hauer,m.,doudna,j.a.,andcharpentier,e.(2012).aprogrammabledual-rna-guideddnaendonucleaseinadaptivebacterialimmunity.science337,816-821.
kagey,m.h.,newman,j.j.,bilodeau,s.,zhan,y.,orlando,d.a.,vanberkum,n.l.,ebmeier,c.c.,goossens,j.,rahl,p.b.,levine,s.s.,etal.(2010).mediatorandcohesinconnectgeneexpressionandchromatinarchitecture.nature467,430-435.
kang,j.y.,song,s.h.,yun,j.,jeon,m.s.,kim,h.p.,han,s.w.,andkim,t.y.(2015).disruptionofctcf/cohesin-mediatedhigh-orderchromatinstructuresbydnamethylationdownregulatesptgs2expression.oncogene34,5677-5684.
konermann,s.,brigham,m.d.,trevino,a.e.,joung,j.,abudayyeh,o.o.,barcena,c.,hsu,p.d.,habib,n.,gootenberg,j.s.,nishimasu,h.,etal.(2015).genome-scaletranscriptionalactivationbyanengineeredcrispr-cas9complex.nature517,583-u332.
laird,p.w.,andjaenisch,r.(1996).theroleofdnamethylationincancergeneticsandepigenetics.annualreviewofgenetics30,441-464.
landau,d.a.,clement,k.,ziller,m.j.,boyle,p.,fan,j.,gu,h.,stevenson,k.,sougnez,c.,wang,l.,li,s.,etal.(2014).locallydisorderedmethylationformsthebasisofintratumormethylomevariationinchroniclymphocyticleukemia.cancercell26,813-825.
lassar,a.b.,paterson,b.m.,andweintraub,h.(1986).transfectionofadnalocusthatmediatestheconversionof10t1/2fibroblaststomyoblasts.cell47,649-656.
li,e.,bestor,t.h.,andjaenisch,r.(1992).targetedmutationofthednamethyltransferasegeneresultsinembryoniclethality.cell69,915-926.
lin,y.,bloodgood,b.l.,hauser,j.l.,lapan,a.d.,koon,a.c.,kim,t.k.,hu,l.s.,malik,a.n.,andgreenberg,m.e.(2008).activity-dependentregulationofinhibitorysynapsedevelopmentbynpas4.nature455,1198-1204.
lister,r.,mukamel,e.a.,nery,j.r.,urich,m.,puddifoot,c.a.,johnson,n.d.,lucero,j.,huang,y.,dwork,a.j.,schultz,m.d.,etal.(2013).globalepigenomicreconfigurationduringmammalianbraindevelopment.science341,629-+.
lister,r.,pelizzola,m.,dowen,r.h.,hawkins,r.d.,hon,g.,tonti-filippini,j.,nery,j.r.,lee,l.,ye,z.,ngo,q.m.,etal.(2009).humandnamethylomesatbaseresolutionshowwidespreadepigenomicdifferences.nature462,315-322.
maeder,m.l.,angstman,j.f.,richardson,m.e.,linder,s.j.,cascio,v.m.,tsai,s.q.,ho,q.h.,sander,j.d.,reyon,d.,bemstein,b.e.,elal.(2013).targeteddnademethylationandactivationofendogenousgenesusingprogrammabletale-tet1fusionproteins.natbiotechnol31,1137-1142.
mali,p.,yang,l.,esvelt,k.m.,aach,j.,guell,m.,dicarlo,j.e.,norville,j.e.,andchurch,g.m.(2013).rna-guidedhumangenomeengineeringviacas9.science339,823-826.
martinowich,k.,hattofi,d.,wu,h.,fouse,s.,he,f.,hu,y.,fan,g.,andsun,y.e.(2003).dnamethylation-relatedchromatinremodelinginactivity-dependentbdnfgeneregulation.science302,890-893.
narendra,v.,rocha,p.p.,an,d.,raviram,r.,skok,j.a.,mazzoni,e.o.,andreinberg,d.(2015).ctcfestablishesdiscretefunctionalchromatindomainsatthehoxclustersduringdifferentiation.science347,1017-1021.
nora,e.p.,lajoie,b.r.,schulz,e.g.,giorgetti,l.,okamoto,i.,servant,n.,piolot,t.,vanberkum,n.l.,meisig,j.,sedat,j.,etal.(2012).spatialpartitioningoftheregulatorylandscapeofthex-inactivationcentre.nature485,381-385.
phillips-cremins,j.e.,sauria,m.e.,sanyal,a.,gerasimova,t.i.,lajoie,b.r.,bell,j.s.,ong,c.t.,hookway,t.a.,guo,c.,sun,y.,etal.(2013).architecturalproteinsubclassesshape3dorganizationofgenomesduringlineagecommitment.cell153,1281-1295.
phillips,j.e.,andcorces,v.g.(2009).ctcf:masterweaverofthegenome.cell137,1194-1211.
qi,l.s.,larson,m.h.,gilbert,l.a.,doudna,j.a.,weissman,j.s.,arkin,a.p.,andlim,w.a.(2013).repurposingcrisprasanrna-guidedplatformforsequence-specificcontrolofgeneexpression.cell152,1173-1183.
robertson,k.d.(2005).dnamethylationandhumandisease.natrevgenet6,597-610.
schultz,m.d.,he,y.,whitaker,j.w.,hariharan,m.,mukamel,e.a.,leung,d.,rajagopal,n.,nery,j.r.,urich,m.a.,chen,h.,etal.(2015).humanbodyepigenomemapsrevealnoncanonicaldnamethylationvariation.nature523,212-216.
seitan,v.c.,faure,a.j.,zhan,y.,mccord,r.p.,lajoie,b.r.,ing-simmons,e.,lenhard,b.,giorgetti,l.,heard,e.,fisher,a.g.,etal.(2013).cohesin-basedchromatininteractionsenableregulatedgeneexpressionwithinpreexistingarchitecturalcompartments.genomeres23,2066-2077.
smith,z.d.,andmeissner,a.(2013).dnamethylation:rolesinmammaliandevelopment.natrevgenet14,204-220.
sofueva,s.,yaffe,e.,chan,w.c.,georgopoulou,d.,vietrirudan,m.,mira-bontenbal,h.,pollard,s.m.,schroth,g.p.,tanay,a.,andhadjur,s.(2013).cohesin-mediatedinteractionsorganizechromosomaldomainarchitecture.emboj32,3119-3129.
stelzer,y.,shivalila,c.s.,soldner,f.,markoulaki,s.,andjaenisch,r.(2015).tracingdynamicchangesofdnamethylationatsingle-cellresolution.cell163,218-229.
sweatt,j.d.(2013).theemergingfieldofneuroepigenetics.neuron80,624-632.
tang,z.,luo,o.j.,li,x.,zheng,m.,zhu,j.j.,szalaj,p.,trzaskoma,p.,magalska,a.,wlodarczyk,j.,ruszczycki,b.,etal.(2015).ctcf-mediatedhuman3dgenomearchitecturerevealschromatintopologyfortranscription.cell163,1611-1627.
vojta,a.,dobrinic,p.,tadic,v.,bockor,l.,korac,p.,julg,b.,klasic,m.,andzoldos,v.(2016).repurposingthecrispr-cas9systemfortargeteddnamethylation.nucleicacidsres44,5615-5628.
wang,h.,maurano,m.t.,qu,h.,varley,k.e.,gertz,j.,pauli,f.,lee,k.,canfield,t.,weaver,m.,sandstrom,r.,etal.(2012).widespreadplasticityinctcfoccupancylinkedtodnamethylation.genomeres22,1680-1688.
wilson,m.h.,coates,c.j.,andgeorge,a.l.,jr.(2007).piggybactransposon-mediatedgenetransferinhumancells.molther15,139-145.
wu,h.,luo,j.,yu,h.,rattner,a.,mo,a.,wang,y.,smallwood,p.m.,erlanger,b.,wheelan,s.j.,andnathans,j.(2014a).cellularresolutionmapsofxchromosomeinactivation:implicationsforneuraldevelopment,function,anddisease.neuron81,103-119.
wu,h.,andzhang,y.(2014).reversingdnamethylation:mechanisms,genomics,andbiologicalfunctions.cell156,45-68.
wu,x.,scott,d.a.,kriz,a.j.,chiu,a.c.,hsu,p.d.,dadon,d.b.,cheng,a.w.,trevino,a.e.,konermann,s.,chen,s.,etal.(2014b).genome-widebindingofthecrisprendonucleasecas9inmammaliancells.natbiotechnol32,670-676.
xu,w.,yang,h.,liu,y.,yang,y.,wang,p.,kim,s.h.,ito,s.,yang,c.,wang,p.,xiao,m.t.,etal.(2011).oncometabolite2-hydroxyglutarateisacompetitiveinhibitorofalpha-ketoglutarate-dependentdioxygenases.cancercell19,17-30.
xu,x.,tao,y.,gao,x.,zhang,l.,li,x.,zou,w.,ruan,k.,wang,f.,xu,g.l.,andhu,r.(2016).acrispr-basedapproachfortargeteddnademethylation.celldiscovery2,16009.
yu,m.,hon,g.c.,szulwach,k.e.,song,c.x.,zhang,l.,kim,a.,li,x.,dai,q.,shen,y.,park,b.,etal.(2012).base-resolutionanalysisof5-hydroxymethylcytosineinthemammaliangenome.cell149,1368-1380.
zhang,y.,liu,t.,meyer,c.a.,eeckhoute,j.,johnson,d.s.,bernstein,b.e.,nusbaum,c.,myers,r.m.,brown,m.,li,w.,etal.(2008).model-basedanalysisofchip-seq(macs).genomebiol9,r137.
zuin,j.,dixon,j.r.,vanderreijden,m.i.,ye,z.,kolovos,p.,brouwer,r.w.,vandecorput,m.p.,vandewerken,h.j.,knoch,t.a.,van,i.w.f.,etal.(2014).cohesinandctcfdifferentiallyaffectchromatinarchitectureandgeneexpressioninhumancells.procnatlacadsciusa111,996-1001.
pctapplicationno.pct/us2014/034387,filedapril16,2014,andu.s.applicationno.15/078,851,filedmarch23,2016.
- 还没有人留言评论。精彩留言会获得点赞!