一种N-芳基萘并呋喃酮亚胺类化合物及其合成方法与流程
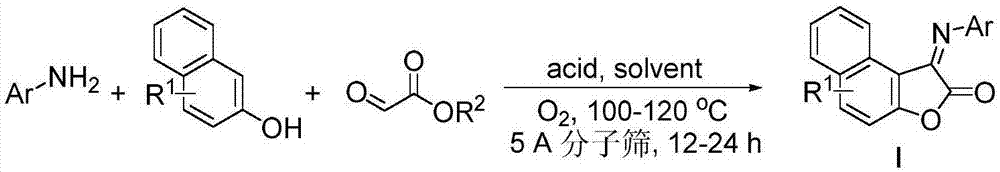
本发明涉及有机合成领域,具体涉及一种n-芳基萘并呋喃酮亚胺类化合物及其合成方法。
【背景技术】
苯并/萘并呋喃酮类化合物及其衍生物的分子骨架大量存在于天然产物、生物活性物质以及功能材料中,特别是有机合成染料中(j.nat.prod.2004,67,882-885;org.lett.2007,9,2449–2451;chem.-eur.j.2008,14,376–381;eur.j.org.chem.2006,102,3–1033;bioorg.med.chem.lett.2007,17,6354–6363)。20世纪七十年代,英国帝国化学工业公司率先推出苯并呋喃酮结构的染料,该类染料具有优异的染色性能且结构完全不同于传统的杂多稠环发色体系,受到了染料界的广泛关注(dyespigm,1980,1,103-120;gb:2068402,1981-08-12;ep:371223,1990-06-06)。
苯胺类染料具有色泽鲜艳、耐洗、发色值高等特点,因而一直是合成染料的主流。将苯胺结构与苯并呋喃酮结构进行结合,衍生出具有全新结构和性能的n-芳基萘并呋喃酮亚胺类化合物则是染料设计的一个新思路。1991年,hafez,taghrid.s.等首先提出使用萘并呋喃-1,2-二酮与邻苯二胺经过脱水缩合得到具有该类骨架的产物(phosphorus,sulfurandsiliconandtherelatedelements,1991,61,341-349)。该种方法需要使用具有强烈致癌作用的苯作为溶剂,且使用的萘并呋喃-1,2-二酮原料需要提前制备,更重要的是,该种方法中仅涉及到1-(2-氨基苯基亚胺)萘并[2,1-b]呋喃-2-酮的合成,对于其它类似结构的产物,该文中并未提出具有相同的适用性。2000年feiler,leonhard等提出使用苯酚或2-萘酚类化合物、草酰氯首先经过傅-克反应发生环合形成1,2-二酮类化合物再与邻氨基苯酚发生分子间脱水缩合,总共历经三步合成得到具有该类骨架的染料(pctint.appl.,2000053597,2000-09-14)。其合成路径与1991年hafez,taghrid.s.等提出的类似,且反应中草酰氯、三氯化铝的使用要求反应必须在严格的无水条件下进行,此外反应中释放出的氯化氢也需要额外的碱与其中和,另外在脱水缩合形成亚胺结构过程中需要使用冰醋酸作为溶剂使得反应条件更加苛刻,最重要的是对于亚胺芳基上除了邻位羟基以外的其它取代基团,该文中也并未提出具有相同的适用性。
针对上述方法的不足,设计具有不同基团取代的n-芳基萘并呋喃酮亚胺类化合物以及开发一种反应条件简单、对环境友好且不需要使用苯或者冰醋酸作为溶剂的方法合成n-芳基萘并呋喃酮亚胺类化合物在染料合成方面是非常重要的,具有巨大的应用前景。
技术实现要素:
本发明的目的是设计开发一系列新型n-芳基萘并呋喃酮亚胺类化合物,以及开发一种反应条件简单、对环境友好且不需要使用苯或者冰醋酸作为溶剂的合成n-芳基萘并呋喃酮亚胺类化合物的方法。
本发明的发明目的是通过如下技术方案实现的:
一种n-芳基萘并呋喃酮亚胺类化合物的合成方法,包含下述步骤:
芳香胺类化合物、2-萘酚类化合物、乙醛酸酯、酸添加剂、
优选地,所述芳香胺类化合物选自苯胺、邻甲基苯胺、邻甲氧基苯胺、间甲基苯胺、间甲氧基苯胺、对甲基苯胺、对甲氧基苯胺、对乙氧基苯胺、对苯氧基苯胺、对叔丁基苯胺、对苯基苯胺、对硫甲基苯胺、n,n-二甲基对苯二苯胺、n,n-二乙基对苯二苯胺、n-苯基对苯二胺、对乙炔基苯胺、邻氯苯胺、间氯苯胺、对氯苯胺、对氟苯胺、对溴苯胺、对硝基苯胺、对三氟甲基苯胺、对苯二胺、对氨基苯酚、对氨基苯甲酸乙酯、对氨基苯乙酮、对氨基苄醇、对氨基苄腈、对氨基苯腈、间氨基吡啶、1-萘胺、2-萘胺、3,5-二甲基苯胺、3,5-二甲氧基苯胺、2,4,6-三甲基苯胺、3-氯-4-甲氧基苯胺。
优选地,所述2-萘酚类化合物选自2-萘酚、6-甲氧基-2-萘酚、7-甲氧基-2-萘酚、7-苯基-2-萘酚、6-溴-2-萘酚、7-溴-2-萘酚、6-羟基-2-萘甲酸、6-羟基-2-萘甲酸甲酯。
优选地,所述乙醛酸酯选自乙醛酸甲酯、乙醛酸乙酯、乙醛酸异丙酯、乙醛酸叔丁酯酯、乙醛酸苯酯。
优选地,所述酸添加剂选自乙酸、叔戊酸、苯甲酸中的至少一种。
优选地,所述有机溶剂选自甲苯、1,2-二氯乙烷、氯苯、二甲苯中的至少一种。
优选地,所述酸添加剂、2-萘酚类化合物、乙醛酸酯、芳香胺类化合物之间的摩尔比为[2~3]:[1~1.5]:[1~1.5]:1。
本发明还提供了上述合成方法制备的苯并/萘并呋喃酮芳基亚胺类化合物,结构式为:
所述结构式i中,
r1是氢、甲氧基、苯基、溴、羧基或甲酸甲酯基;
ar是苯基、邻甲基苯基、邻甲氧基苯基、间甲基苯基、间甲氧基苯基、对甲基苯基、对甲氧基苯基、对乙氧基苯基、对苯氧基苯基、对叔丁基苯基、对联苯基、对硫甲基苯基、对n,n-二甲基苯基、对n,n-二乙基苯基、对n-苯基苯基、对乙炔基苯基、邻氯苯基、间氯苯基、对氯苯基、对氟苯基、对溴苯基、对硝基苯基、对三氟甲基苯基、对氨基苯基、对羟基苯基、对甲酸乙酯基苯基、对乙酰基苯基、对甲醇基苯基、对乙腈基苯基、对氰基苯基、间吡啶基、1-萘基、2-萘基、3,5-二甲基苯基、3,5-二甲氧基苯基、2,4,6-三甲基苯基、3-氯-4-甲氧基苯基;
其中,结构式i中r1是定位在7-,8-位中的至少一个位置。
根据实验结果,本发明提供了一种新型n-芳基萘并呋喃酮亚胺类化合物,以及在氧气氧化的情况下使用芳香胺类化合物、2-萘酚类化合物以及乙醛酸酯一锅法合成n-芳基萘并呋喃酮亚胺类化合物的新方法。该方法具有官能团兼容性强(如烷基、烷氧基、芳基、含硫含氮的基团、卤素、硝基、三氟甲基、酯基、羟基、乙酰基、炔基、杂环)、反应条件简单、所得目标产物易分离、产率较高等特点。该方法避免了苯或者冰醋酸作为溶剂以及草酰氯作为酰化试剂的使用,解决了苯、冰醋酸对环境危害以及需要额外添加碱等问题,并且该一锅法制备以及氧气作为氧化剂的使用为该新型n-芳基萘并呋喃酮亚胺类化合物的合成提供了一种更加经济环保的路径。
【附图简要说明】
图1为一种n-芳基萘并呋喃酮亚胺类化合物合成的反应式。
【具体实施方式】
下面结合本发明的合成例对本发明所述的合成方法作进一步说明,需要说明的是,实施例并不构成对本发明要求保护范围的限制。
如图1所示,本发明提供的n-芳基萘并呋喃酮亚胺类化合物合成步骤为:将芳香胺类化合物、2-萘酚类化合物(摩尔比100%-150%基于芳香胺类化合物)、乙醛酸酯(摩尔比100%-150%基于芳香胺类化合物)、酸添加剂(摩尔比200%-300%基于芳香胺类化合物)、
合成例1
(z)-1-(4-苯氨基苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmoln-苯基对苯二胺,0.3mmol2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例2
(z)-1-(1-萘基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol1-萘胺,0.3mmol2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例3
(z)-1-(4-联苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol对苯基苯胺,0.3mmol2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例4
(z)-1-(3,5-二甲基苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol3,5-二甲基苯胺,0.3mmol2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例5
(z)-1-(3-氯-4-甲氧基苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol3-氯-4-甲氧基苯胺,0.3mmol2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例6
(z)-8-甲氧基-1-(4-氟苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol对氟苯胺,0.3mmol7-甲氧基-2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例7
(z)-8-甲氧基-1-(4-氯苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol对氯苯胺,0.3mmol7-甲氧基-2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例8
(z)-8-甲氧基-1-(4-溴苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol对溴苯胺,0.3mmol7-甲氧基-2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例9
(z)-8-甲氧基-1-(4-三氟甲基苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol对三氟甲基苯胺,0.3mmol7-甲氧基-2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
合成例10
(z)-8-甲氧基-1-(4-乙炔基苯基亚胺)萘并[2,1-b]呋喃-2-酮
在25ml充满氧气的反应器中加入0.2mmol对乙炔基苯胺,0.3mmol7-甲氧基-2-萘酚、0.2mmol乙醛酸乙酯,0.4mmol叔戊酸、
- 还没有人留言评论。精彩留言会获得点赞!