一种基于丙炔基成醚的酚羟基保护方法与流程
本发明属于化学合成技术领域,具体涉及一种基于丙炔基成醚的酚羟基保护方法。
背景技术
在有机合成中,酚羟基是一种重要的官能团,其本身具有功能性,通过化学反应可将其转变为其他的官能团,酚羟基比较活泼,其与氧化剂和亲电试剂等极易发生反应,一个亲核的酚氧离子即使是与弱的烷基化试剂或者酰基化试剂也易发生反应,所以在多步有机合成反应中,需要用保护基将其保护起来,在合适的时候再脱去保护基以利于后续反应的顺利进行。
其中,甲醚基是酚羟基最常用的保护基,因为甲醚基不但容易制备,而且对一系列试剂稳定;但是,用甲醚基保护酚羟基存在一个问题,由于酚甲醚不受酸、碱、氧化剂、还原剂和亲核试剂的影响,所以脱去甲醚保护基时需要在剧烈的条件下进行,反应进程不易控制。
常用的脱酚羟基保护基的方法有:三氯化铝法、氢溴酸法、三溴化硼法等。据a.m.bernard,m.r.ghiani,p.p.piras.anda.rivoldini.synthesis,287(1989)文献报道,在dmf中用氯化锂可以选择性的脱去甲基。
但按上述方法脱去酚羟基甲基保护基的反应比较剧烈,不易控制反应的进程,因此,找到温和的脱保护基的试剂和方法很有必要。
技术实现要素:
针对上述存在的问题,本发明旨在提供一种新的高效的基于丙炔基成醚的酚羟基保护方法,丙炔基保护的苯基醚结构稳定,在多步合成反应中有利于后续反应的进行;脱保护条件温和,可以完全去除保护基,且不影响其他苯基烷烃醚结构,副产物少、产率高、操作简单。
为了实现上述目的,本发明所采用的技术方案如下:一种基于丙炔基成醚的酚羟基保护方法,具体反应路线如下:
其中,r1和r2为烷基、烷氧基、芳香基或酰基。
进一步地,本方法的具体的反应过程为:
保护反应:溴丙炔首先与含有酚羟基的底物反应,生成丙炔基保护的苯基醚;
脱保护反应:脱保护时以醇类做溶剂,将30-100eq的醋酸铵加入到含有与醋酸氨当量比的原料的反应体系中,回流条件下反应4-13h,得到酚羟基脱保护后的目标物质。
进一步地,所述底物包括一元酚和多元酚及其衍生物,底物中的芳香环结构包括单环结构和多环结构。
进一步地,进行脱保护反应时所用的醇类溶剂优选为正丁醇溶剂,正丁醇的用量为每1.0g反应原料中添加20~50ml正丁醇。
进一步地,进行脱保护反应时的醋酸铵用量优选为50-60eq。
进一步地,进行脱保护反应时的回流反应温度为70-150℃。
进一步地,进行脱保护反应时的回流反应温度优选为135-140℃。
本发明的有益效果是:本发明公开的一种基于丙炔基成醚的酚羟基保护方法是以溴丙炔与底物中的酚羟基进行反应以生成丙炔基保护的苯基醚,该基团结构稳定,有利于多步化学合成反应中后续反应的进行;脱保护反应的条件简单,可以在不破坏其他苯基烷烃醚的基础上完全去除特定结构的酚羟基底物上的丙炔基保护基,特异性较强;本发明公开的方法不仅反应条件温和,反应进程容易掌控,而且副产物少,产率高。
附图说明
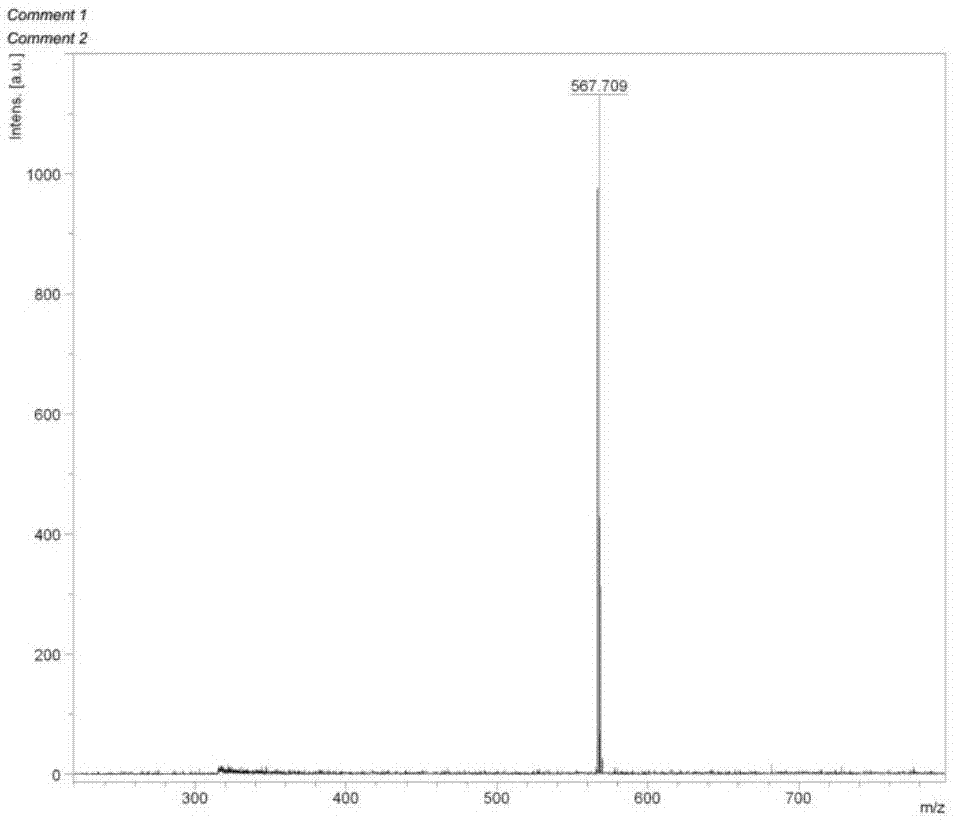
图1为本发明实施例1制备的化合物4的质谱图;
图2为本发明实施例2制备的化合物7的质谱图。
具体实施方式
为了使本领域的普通技术人员能更好的理解本发明的技术方案,下面结合附图和实施例对本发明的技术方案做进一步的描述。
需要说明的是,所描述的实施例仅仅是本发明的部分较佳实施例。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例,相反地,提供这些实施例的目的是为了技术人员能对本发明公开内容的理解更加透彻全面。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例都应看作是本发明的简单变型,都在本发明的保护范围内。
实施例一:该实施例中具体公开的制备路线包括对酚羟基进行保护的步骤,具体的反应路线为:
化合物1的合成
将2.73g的对羟基苯乙酮(约20mmol)、3.54g的溴丙炔(约30mmol)、4.15g碳酸钾加入到50ml的dmf溶液中,在磁力搅拌下,80℃反应24h;反应结束后,水/二氯甲烷萃取多次,合并有机相;经层析柱分离,得到黄色固体2.88g(产率约60%)即化合物1。
1hnmr(400mhz,cdcl3)δ7.97(d,j=8.8hz,2h),7.04(d,j=8.8hz,2h),5.32(s,3h),4.79(s,2h),1.68(s,1h).
化合物2的合成
将2.61g的化合物1(约15mmol)、2.24g的对二甲氨基苯甲醛(约15mmol)加入到40ml的乙醇溶液中,搅拌至固体全部溶解后,缓慢加入5ml氢氧化钾溶液(含1.00g氢氧化钾),于室温下搅拌24h,反应过程中有黄色固体析出;反应结束后,用1m的盐酸溶液调节反应液至中性,抽滤得固体,去离子水洗涤三次,真空干燥得6.50g的黄色固体(产率约97%)即化合物2。
1hnmr(400mhz,cdcl3)δ8.06(d,j=8.0hz,2h),7.82(d,j=12.0hz,1h),7.57(d,j=8.0hz,2h),7.37(d,j=16.0hz,1h),7.08(d,j=12.0hz,2h),6.72(d,j=8.0hz,2h),4.80(s,2h),3.07(t,,j=16.0hz,6h),2.59(t,j=4hz,1h).
化合物3的合成
在氮气保护下加入3.05g的化合物2(约10mmol)、2.75g的硝基甲烷(约50mmol)、3.9g的二乙胺(约50mmol)和50.0ml的无水甲醇,加热回流24h后,用1m的盐酸溶液调节反应液至中性,抽滤得固体,冷甲醇洗涤三次,真空干燥后得到2.56g的产品(产率约70%)即化合物3。
1hnmr(400mhz,dmso)δ7.81(d,j=8.0hz,2h),7.14(d,j=8.0hz,2h),6.83(d,j=8.0hz,2h),6.62(d,j=8.0hz,2h),4.90(dd,j=8.0,16.0hz,1h),4.75(dd,j=12.0,16.0hz,1h),3.40-3.30(m,5h),2.84(s,6h).
化合物4的合成
将2.20g(6.00mmol)的化合物3、21.48g(约300mmol)的醋酸铵固体加入到60ml正丁醇溶液中,磁力搅拌下,加热回流约12h,减压浓缩到原来体积的1/4,待溶液冷却到室温,过滤得到褐色固体,无水乙醇洗涤,干燥后得到1.12g蓝色固体产物(产率约33%)即化合物4。
对化合物4进行maldi–tof测定,测定结果如图1,[m/e](m,maldi-tof)理论值:567.26,实验值:567.71。
实施例二:该实施例中具体公开的制备路线同样包括对酚羟基进行保护的过程,具体的反应路线为:
其中,化合物1的合成方法与实施例一中相同;
化合物5的合成
将2.61g的化合物1(约15mmol)、2.04g的对甲氧基苯甲醛(约15mmol)加入到40ml的乙醇溶液中,搅拌至固体全部溶解后,缓慢加入5ml氢化钾溶液(含1.00g氢氧化钾),于室温下搅拌反应24h,反应过程中有黄色固体析出,反应结束后,用1m的盐酸溶液调节反应液至中性,抽滤得固体,去离子水洗涤三次,真空干燥得4.25g的黄色固体(产率约97%)即化合物5。
1hnmr(400mhz,cdcl3)δ8.07(d,j=8.0hz,2h),7.81(d,j=16.0hz,1h),7.63(d,j=8.0hz,2h),7.45(d,j=16.0hz,1h),7.09(d,j=8.0hz,2h),6.96(d,j=8.0hz,2h),4.81(d,j=4.0hz,2h),3.89(s,3h),2.59(s,1h).
化合物6的合成
在氮气保护下加入2.92g的化合物5(约10mmol)、2.75g的硝基甲烷(约50mmol)、3.9g的二乙胺(约50mmol)和50.0ml的无水甲醇,加热回流24h后,用1m的盐酸溶液调节反应液至中性,抽滤得固体,冷甲醇洗涤三次,真空干燥后得到2.47g的产品(产率约70%)即化合物6。
1hnmr(400mhz,cdcl3)δ7.84(d,j=8.0hz,2h),7.20(d,j=8.0hz,2h),6.86(dd,j=8.0,8.0hz,4h),4.80(dd,j=8.0,12.0hz,1h),4.65(dd,j=8.0,12.0hz,1h),4.20–4.11(m,1h),3.77(s,3h),3.44-3.11(m,2h),1.29(s,2h),0.92-0.89(m,1h).
化合物7的合成
将2.12g(6.00mmol)的化合物6、21.48g(约300mmol)醋酸铵固体加入到60ml正丁醇溶液中,磁力搅拌下加热回流约12h,减压浓缩到原来体积的1/4,待溶液冷却到室温,过滤得到褐色固体,无水乙醇洗涤,干燥后得到1.07g蓝色固体产物(产率约33%)即化合物7。
对化合物7进行maldi–tof测定,结果如图2,[m/e](m,maldi-tof)理论值:541.20,实验值:541.63。
从上述两个实施例的实验结果可以看出,本发明公开的方法在多步化合反应中能够成功保护酚羟基并得到了目标物质,未对其他基团造成影响,且全程反应条件温和,反应进程易于控制,操作简单,副产物少。
以上显示和描述了本发明的基本原理、主要特征及优点。但是以上所述仅为本发明的具体实施例,本发明的技术特征并不局限于此,任何本领域的技术人员在不脱离本发明的技术方案下得出的其他实施方式均应涵盖在本发明的专利范围之中。
- 还没有人留言评论。精彩留言会获得点赞!