萘霉素类化合物及其制备方法与其应用与流程
本发明属于医药技术领域,具体涉及一种利用链霉菌的发酵液制备的六种萘霉素类新化合物及其制备方法与其应用,经结构鉴定,本发明六种化合物均为新化合物,分别命名为化合物1、2、3、4、5和6。
技术背景
安莎霉素(ansamycins)类化合物是一类主要由微生物产生的结构较复杂的大环内酰胺类抗生素。这类化合物的结构特征是由一平面的芳香核和连接芳香核两个不相邻位置的脂肪链“桥”所组成的环状结构。安莎霉素类化合物几乎都具有良好的生物活性,其中最具有代表性的是格尔的霉素和美登木素的衍生物用于治疗肿瘤,利福霉素的衍生物用于治疗结核。萘霉素(napthomycin)作为安莎霉素的一个亚族同样具有良好的生物活性,发掘新颖的萘霉素对于发现新药具有重要意义。
技术实现要素:
本发明的目的在于提供一种萘霉素类化合物及其制备方法与其在制药中的应用,特别是在制备抗癌药物中的应用。
为了实现本发明的上述目的,本发明提供了如下的技术方案:
如下结构式所示的萘霉素类化合物,
六个新化合物的制备方法,包括下列步骤:
1.发酵
(1)发酵菌株:发酵菌株为streptomyceskib-h2054,由东北农业大学生物化工实验室提供。
(2)平面培养:采用ms培养基(大豆粉20g/l,甘露醇20g/l,琼脂粉20g/l,ph=7.0~7.2,使用去离子水配制和定容,于121℃下灭菌30分钟),接种后在28℃下培养5天。
(3)种子培养:种子培养基(胰蛋白胨17.0g/l,植物蛋白胨3.0g/l,氯化钠5.0g/l,磷酸氢二钾2.5g/l,葡萄糖2.5g/l,ph7.0~7.2,使用去离子水配制和定容,于121℃下灭菌30分钟),用250ml的摇瓶每瓶分装50ml,然后用竹签刮取平面的孢子接种至摇瓶,置于摇床上28℃,200rpm培养24-36小时。
(4)发酵:采用液体摇瓶发酵,发酵培养基(可溶性淀粉10.0g/l,胰蛋白胨5.0g/l,葡萄糖10.0g/l,甘油10.0g/l,酵母提取粉5.0g/l,caco33g/l,使用去离子水配制和定容,于121℃下灭菌30分钟),用1l的摇瓶每瓶分装250ml,然后按照5%v/v的接种量将预培养的种子液接种至摇瓶中,于28℃,200rpm培养5天。
2.发酵液处理:
20l发酵液经离心(4000rpm,25℃),分成菌丝体和上清液;两部分,菌丝体用丙酮浸泡超声提取后,减压蒸馏除去丙酮,以等体积乙酸乙酯萃取三遍,减压蒸馏浓缩得菌丝体浸膏。上清液用等体积乙酸乙酯萃取三遍,减压蒸馏浓缩得上清液浸膏。经hplc分析两部分浸膏的成分后,基于相似的主要成分合并两部分浸膏。
3.化合物制备
(1)将粗提浸膏用60-100目的硅胶采用干法装柱的方式进行出分离,用体积比为1:0-1:1的石油醚-乙酸乙酯溶液进行梯度洗脱,得组分i-vi;纯液乙酸乙酯洗脱的组分(v)经凝胶柱色谱sephadexlh-20,甲醇洗脱,得组分v1-v6;将组分v2经半制备hplc(色谱柱ymctriartc1810×250mm,5μm),使用50%乙腈水溶液(0.1%乙酸)的流动相进行洗脱得到化合物1和2;将组分v3经c18半制备柱,85%甲醇水溶液(0.1%乙酸)洗脱得化合物3和4;将组分v4经c18半制备柱,52%乙腈水溶液(0.1%乙酸)洗脱得化合物napthomycine。
(2)通过将5mg的napthomycine溶于1ml甲苯,在254nm的紫外光下照射30分钟,经c18半制备柱,使用80%甲醇(0.1%乙酸)的流动相进行洗脱得到所需化合物5和6。
如所述的萘霉素类化合物的制备方法,所述的紫外光的波长为254nm。
如所述的萘霉素类化合物的制备方法,所述的高效液相色谱法分离纯化采用250mm×10mm的c18半制备柱,流速为3.0ml/min。
一种治疗癌症的药物,包括治疗有效量的萘霉素类化合物,和至少一种药学上可接受的载体。
所述的萘霉素类化合物在制备治疗癌症的药物中的应用。
所述的萘霉素类化合物在制备治疗白血病、肝癌、肺癌、乳腺癌和结肠癌的药物中的应用。
与现有技术相比,本发明具有以下有益效果:
1.本发明所述的技术方案可以为将来这六个新化合物开发成治疗人类癌症的药物解决原料来源问题,该技术具有培养条件温和、培养基配制简单易行、发酵时间短、产量大、易于大规模生产等优点。
2.分离得到的化合物细胞毒活性实验表明,化合物1只对白血病(hl-60)有较好的细胞毒活性ic50值为18.4μm,表明其对五株测试细胞株中的hl-60细胞株有较强的选择性细胞毒活性。化合物3和化合物5对五株测试细胞株均具有较好的细胞毒活性ic50值为10~20μm,尤其是化合物3对乳腺癌细胞(mcf-7)的ic50值达到10.7μm,为后续应用开发奠定了科学基础。
附图说明
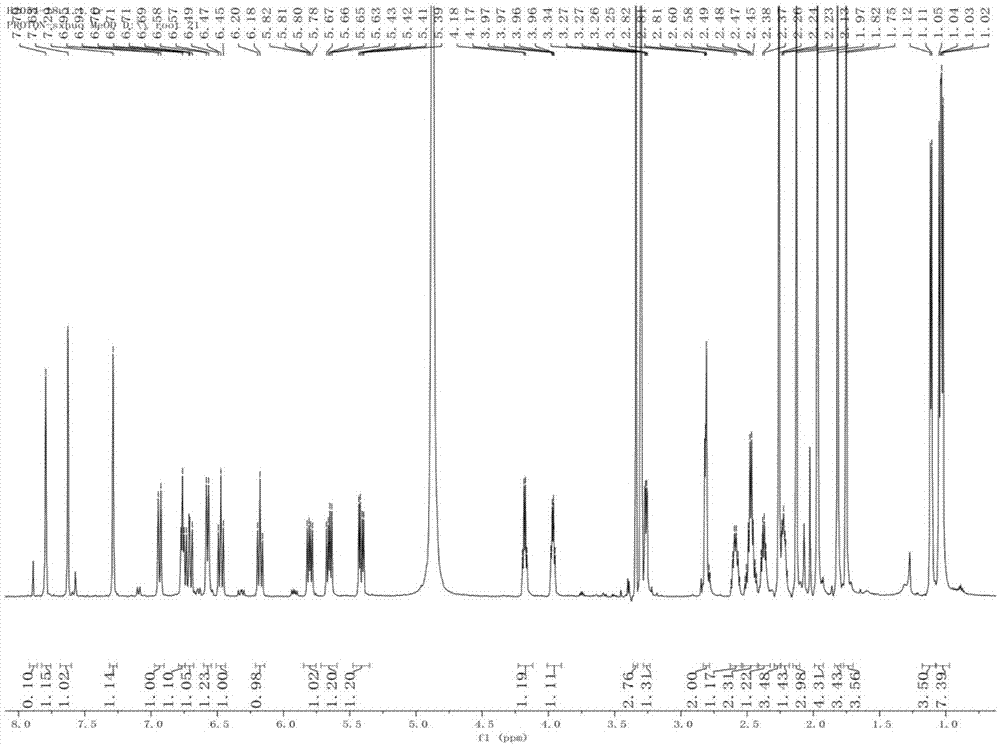
图1为本发明的化合物1的1hnmr谱图。
图2为本发明的化合物1的13cnmr谱图。
图3为本发明的化合物2的1hnmr谱图。
图4为本发明的化合物2的13cnmr谱图。
图5为本发明的化合物3的1hnmr谱图。
图6为本发明的化合物3的13cnmr谱图。
图7为本发明的化合物4的1hnmr谱图。
图8为本发明的化合物4的13cnmr谱图。
图9为本发明的化合物5的1hnmr谱图。
图10为本发明的化合物5的13cnmr谱图。
图11为本发明的化合物6的1hnmr谱图。
图12为本发明的化合物6的13cnmr谱图。
图13为本发明6个新化合物的结构示意图。
具体实施方式
为使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。但并不以此来限定本发明。
实施例1
本发现的目的在于提供一种利用链霉菌的发酵液制备化合物1-6的方法。
为实现上述目的,本发现采取的技术方案为:
1.发酵
(1)发酵菌株:发酵菌株为streptomyceskib-h2054,由东北农业大学生物化工实验室提供。
(2)平面培养:采用ms培养基(大豆粉20g/l,甘露醇20g/l,琼脂粉20g/l,ph=7.0~7.2,使用去离子水配制和定容,于121℃下灭菌30分钟),接菌后在28℃下培养5天。
(3)种子培养:种子培养基(胰蛋白胨17.0g/l,植物蛋白胨3.0g/l,氯化钠5.0g/l,磷酸氢二钾2.5g/l,葡萄糖2.5g/l,ph7.0~7.2,使用去离子水配制和定容,于121℃下灭菌30分钟),用250ml的摇瓶每瓶分装50ml,然后用竹签刮取平面的孢子接种至摇瓶,置于摇床上28℃,200rpm培养24-36小时。
(4)发酵:采用液体摇瓶发酵,发酵培养基(可溶性淀粉10.0g/l,胰蛋白胨5.0g/l,葡萄糖10.0g/l,甘油10.0g/l,酵母提取粉5.0g/l,caco33g/l,使用去离子水配制和定容,于121℃下灭菌30分钟),用1l的摇瓶每瓶分装250ml,然后按照5%v/v的接种量将预培养的种子液接种至摇瓶中,于28℃,200rpm培养5天。
2.发酵液处理:
20l发酵液经离心(4000rpm,25℃),分成菌丝体和上清液;两部分,菌丝体用丙酮浸泡超声提取后,减压蒸馏除去丙酮,以等体积乙酸乙酯萃取三遍,减压蒸馏浓缩得菌丝体浸膏。上清液用等体积乙酸乙酯萃取三遍,减压蒸馏浓缩得上清液浸膏。经hplc分析两部分浸膏的成分后,基于相似的主要成分合并两部分浸膏。
3.化合物制备
(1)化合物制备:将粗提浸膏用60-100目的硅胶采用干法装柱的方式进行出分离,用体积比为1:0-1:1的石油醚-乙酸乙酯溶液进行梯度洗脱,得组分i-vi;纯液乙酸乙酯洗脱的组分(v)经凝胶柱色谱sephadexlh-20,甲醇洗脱,得组分v1-v6;将组分v2经半制备hplc(色谱柱ymctriartc1810×250mm,5μm),使用50%乙腈水溶液(0.1%乙酸)的流动相进行洗脱得到化合物1和2;将组分v3经c18半制备柱,85%甲醇水溶液(0.1%乙酸)洗脱得化合物3和4;将组分v4经c18半制备柱,52%乙腈水溶液(0.1%乙酸)洗脱得化合物napthomycine。
(2)通过将5mg的napthomycine溶于1ml甲苯,在254nm的紫外光下照射30分钟,经c18半制备柱,使用80%甲醇(0.1%乙酸)的流动相进行洗脱得到所需化合物5和6。
实施例2
对所纯化的化合物1-6进行表征,表征内容如下:旋光度、紫外光谱(甲醇溶剂)、红外光谱(溴化钾压片)、高分辨质谱、1d和我2d核磁共振谱,所采用的表征方法均按照标准操作,无任何修改。
化合物1:旋光
化合物2:旋光
表1化合物1和2的1hnmr和13cnmr的数据
化合物3:旋光
化合物4:旋光
表2化合物3和4的1hnmr和13cnmr的数据
化合物5:旋光
化合物6:旋光
表3化合物5和6的1hnmr和13cnmr的数据
实施例3
本发明化合物的细胞毒性和细胞增殖情况的影响分别采用mtt法进行分析评估,分别对人白血病细胞(hl-60)、人肝癌细胞(smmc-7721)、人肺癌细胞(a589)、人乳腺癌细胞(mcf-7)和人结肠癌细胞(sw480)进行细胞毒实验,具体实验方法参照标准实验方案进行,无其他修改。
化合物1对人白血病细胞(hl-60)的ic50值为18.38μm、人肝癌细胞(smmc-7721)的ic50值为32.55μm,对其他三株细胞株的ic50大于40μm,表明其对五株测试细胞株的细胞毒活性具有选择性;化合物3对人肺癌细胞(a589)、人乳腺癌细胞(mcf-7)的毒性较大,ic50值为10.68,对其他四株细胞株的ic50值为15~18μm;化合物5对五株癌细胞株均有一定的细胞毒活性,ic50值为16~20μm。
实施例4:
胶囊剂:化合物1-6分别或混合10mg,乳糖187mg,硬脂酸镁3mg。
制备方法:将化合物1-6分别或混合,与助溶剂混和,过筛,均匀混合,把得到的混合物装入硬明胶胶囊,每个胶囊重200mg,活性成分含量为10mg。
实施例5:
片剂:将化合物1-6分别或混合10mg,加入乳糖180mg,淀粉55mg等辅料混合均匀,并用水将其润湿后制成颗粒并干燥,再加入硬脂酸镁5mg混匀后压片,每片约重250mg。
实施例6:
安瓿剂:将化合物1-6分别或混合2mg,氯化钠10mg,溶解于适量的注射用水中,过滤所得溶液,在无菌条件下装入安瓿瓶中。
实施例10:
注射用冻干剂:将化合物1-6分别或混合10mg,碳酸氢钠2mg,甘露醇252mg。
制备方法:将碳酸氢钠、甘露醇,加注射用水溶解,加活性炭吸附30min除热原,过滤去除活性炭,在滤液中加入化合物或其盐,超声处理使溶解,用1n盐酸调节ph为5.0-7.0,微孔滤膜滤过,加注射用水,分装,冷冻干燥,上塞,轧盖,即得。
实施例7:
胶囊剂:将化合物1-6分别或混合10mg,乳糖187mg,硬脂酸镁3mg。
制备方法:将化合物i或ii或其盐与助溶剂混合,过筛,均匀混合,把得到的混合物装入硬明胶胶囊,每个胶囊重200mg,活性成分含量为10mg。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!