正电子显像用化合物、其中间体、制备方法和显像剂与流程
本发明涉及正电子药物与正电子发射断层显像(positronemissiontomography,pet)的应用领域,具体涉及一类靶向pi3k/akt/mtor信号通路的新型pet分子探针及其制备方法和应用。
背景技术
磷脂酰肌醇-3-激酶(phosphatidylinositol-3-kinase,pi3k)信号转导通路在肿瘤的发生、发展、治疗及转归中发挥重要作用,在乳腺癌、大肠癌、子宫内膜癌、脑癌、胃癌、肺癌等很多恶性肿瘤中均发现了该通路的异常激活(bader,
a.g.,etal.,oncogenicpi3kderegulatestranscriptionandtranslation.natrevcancer,2005.5(12):p.921-9.)。pi3k可以激活启动信号转导级联反应,其中最主要的下游激酶是丝/苏氨酸激酶(proteinkinaseb,akt)和哺乳动物类雷帕霉素靶蛋白(mammaliantargetofrapamycin,mtor)(huang,j.andb.d.manning,acomplexinterplaybetweenakt,tsc2andthetwomtorcomplexes.biochemsoctrans,2009.37(pt1):p.217-22.)。
在一项包含19784例实体瘤病人的回顾性研究中,病理证实的pi3k/akt/mtor信号通路的突变率为38%(millis,s.z.,etal.,landscapeofphosphatidylinositol-3-kinasepathwayalterationsacross19784diversesolidtumors.jamaoncol,2016.2(12):p.1565-1573)。在实体瘤中,乳腺癌是女性最常见的恶性肿瘤,其中大约70%的乳腺癌患者存在该通路的突变(cancergenomeatlas,n.,comprehensivemolecularportraitsofhumanbreasttumours.nature,2012.490(7418):p.61-70.)。编码pi3k催化亚基p110α的pik3ca基因的突变,以及人第10号染色体缺失的磷酸酶(tensionhomologydeletedonchromosometen,pten)的缺失,是导致乳腺癌pi3k信号通路突变的主要原因(samuels,y.andt.waldman,oncogenicmutationsofpik3cainhumancancers.currtopmicrobiolimmunol,2010.347:p.21-41)。有报道发现大约有45%原发性乳腺恶性肿瘤存在pik3ca基因突变(外显子9和/或外显子20),而且在实体瘤中,pi3k的表达水平与疾病的预后及疗效评估密切相关(o'brien,c.,etal.,predictivebiomarkersofsensitivitytothephosphatidylinositol3'kinaseinhibitorgdc-0941inbreastcancerpreclinicalmodels.clincancerres,2010.16(14):p.3670-83)。因此,近些年来,该信号通路目前已成为肿瘤治疗的研究热点,很多制药公司和实验室都在积极研发pi3k抑制剂,并有部分已经进入临床试验。
然而,并不是所有的实体瘤患者均存在pik3ca基因突变,要更好的利用pi3k抑制剂进行分子靶向治疗,必须对患者进行必要的筛选,以避免盲目的给药而给病人来带的身体和经济上的负担。在临床工作中,pi3k蛋白的表达水平的检测常常需要对病人的肿瘤组织进行免疫组化检测,而这通常都是有创检查,且检测的准确性与取材密切相关。
随着无创性分子影像成像技术的发展,利用放射性核素示踪技术追踪疾病的早期功能代谢改变展现出了明显的优势,其能够比解剖学显像更早且更全面地反映疾病状况。
目前pet-ct显像的主要显像剂是18f-fdg,即正电子核素18f标记在葡萄糖上的18f-脱氧葡萄糖。18f-fdg可准确反映体内器官/组织的葡萄糖代谢水平,而恶性肿瘤细胞因代谢旺盛,对葡萄糖的需求增加,因而18f-fdg已经广泛应用于肿瘤的检测和诊断中,可早期发现肿瘤原发及转移病灶,准确判断其良、恶性,从而正确指导临床治疗决策。然而,尽管18f-fdgpet已经成功应用于临床肿瘤诊断中,但是18f-fdgpet显像在进一步诊断和筛选患者是否存在pik3ca基因突变而导致肿瘤的应用中难以发挥作用,这是由于pi3k蛋白的表达水平与高摄取葡萄糖并无直接关联。
因此,需要开发一种靶向pi3k通路的显像剂,从而提供一种有效且无创的方法来检测肿瘤患者pi3k蛋白的表达水平,从而进一步筛选出存在pik3ca基因突变的患者以便进行针对性治疗。
技术实现要素:
针对上述情况,本发明提供了靶向pi3k通路的分子探针,用于无创评估患者的pi3k/akt/mtor信号通路表达情况。因此,本发明一方面为pi3k蛋白抑制剂敏感病人进行分子靶向治疗提供有效的筛选工具,另一方面还在于对pi3k/akt/mtor信号通路抑制剂类药物的疗效提供评估工具。
根据本发明的第一方面,提供一种式i或式ii所示的化合物:
其中,r为具有至少一个18f取代基的c1-12的烷基、-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1-10的整数,y为1-5的整数,z为1-3的整数。
根据优选的实施方式,上述式i和式ii中,r为具有至少一个18f取代基的c1-6的烷基、-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1-5的整数,y为1-3的整数,z为1。更优选所述r具有1-3个18f取代基。
进一步优选地,所述具有至少一个18f取代基的c1-6的烷基选自由被1-3个18f取代的甲基、乙基、丙基、丁基、戊基和己基所组成的组;和所述具有至少一个18f取代基的-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3选自端甲基被1-3个18f取代的-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1-5的整数,y为1-3的整数,z为1。
更优选地,r为在一个端甲基上具有一个18f取代基的甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1、2或3,y为1或2,z为1。
根据具体实施方式,r可选自:-c18f3、-ch18f2、-ch218f、-ch2c18f3、-ch2ch18f2、-ch2ch218f、-ch2ch2c18f3、-ch2ch2ch18f2、-ch2ch2ch218f、-ch(ch3)c18f3、-ch(ch3)ch18f2、-ch(ch3)ch218f、-ch2ch2ch2c18f3、-ch2ch2ch2ch18f2、-ch2ch2ch2ch218f、-c(ch3)2c18f3、-c(ch3)2ch18f2、-c(ch3)2ch218f、-ch2och218f、-ch2och2och218f、-ch2och2och2och218f、-ch2ch2och2ch218f、-ch2ch2och2ch2och2ch218f、-ch2ch2ch2och2ch2ch218f,但不限于此。
更优选,r可选自:-ch2och2och218f、-ch2och2och2och218f、-ch2ch2och2ch218f、-ch2ch2och2ch2och2ch218f、-ch2ch2ch2och2ch2ch218f。
最优选,r可选自:-ch2ch2och2ch2och2ch218f。
本发明化合物中,烷氧单元的引入改善了水溶性,使得其在肝脏中的代谢加快,还增加了探针在血液中的滞留时间,有利于探针在肿瘤等靶部位的蓄积,因而相对于链烷基更为优选。
本发明优选的具体化合物为:
r=-ch2ch2och2ch2och2ch218f的化合物:
4-(2-(1-(2-(2-(2-(氟代-18f)乙氧基)乙氧基)乙基)-1h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉(4-(2-(1-(2-(2-(2-(fluoro-18f)ethoxy)ethoxy)ethyl)-1h-indazol-4-yl)-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine),和
4-(2-(1-(2-(2-(2-(氟代-18f)乙氧基)乙氧基)乙基)-2h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉(4-(2-(1-(2-(2-(2-(fluoro-18f)ethoxy)ethoxy)ethyl)-2h-indazol-4-yl)-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine);
r=-ch2ch2och2ch218f的化合物:
4-(2-(1-(2-(2-(氟代-18f)乙氧基)乙基)-1h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉(4-(2-(1-(2-(2-(2-(fluoro-18f)ethoxy)ethoxy)ethyl)-1h-indazol-4-yl)-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine),和
4-(2-(1-(2-(2-(氟代-18f)乙氧基)乙基)-2h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉(4-(2-(1-(2-(2-(2-(fluoro-18f)ethoxy)ethoxy)ethyl)-2h-indazol-4-yl)-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine);
r=-ch2ch218f的化合物:
4-(2-(1-(2-(氟代-18f)乙基)-1h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉(4-(2-(1-(2-(fluoro-18f)ethyl)-1h-indazol-4-yl)-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine),和
4-(2-(1-(2-(氟代-18f)乙基)-2h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉(4-(2-(1-(2-(fluoro-18f)ethyl)-2h-indazol-4-yl)-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine)。
在一个尤其优选的实施方式中,所述化合物为以下化合物1或化合物2:
本发明第二方面提供一种具有以下结构式i’或ii’的中间体:
其中,r’(rm)n表示被n个rm基团取代的c1-12的烷基、-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1-10的整数,y为1-5的整数,z为1-3的整数,n为1或更大的整数,rm是选自由-ots(对甲苯磺酸酯基)、-oms(甲基磺酸酯基)、-otf(三氟甲磺酸酯基),-bf3、-苯基-(r1)q、-i、-br和-cl组成的组中的离去基团,其中r1为no2、n(ch3)3或sn(ch3)2,q为1-5的整数。
根据一种优选实施方式,所述的式i’或ii’的中间体中r’(rm)n表示被n个rm基团取代的c1-6的烷基、-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1-5的整数,y为1-3的整数,z为1,n为1或更大的整数,rm是选自由-ots、-oms、-bf3、-p-n(ch3)3-ph-,-p-no2-ph-、-sn(ch3)3-ph-、-i、-br和-cl组成的组中的离去基团。优选地,n为1-3的整数。
进一步优选地,所述被n个rm基团取代的c1-6的烷基选自由被1-3个rm基团取代的甲基、乙基、丙基、丁基、戊基和己基所组成的组;和所述被n个rm基团取代的-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3选自端甲基被1-3个rm基团取代的-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1-5的整数,y为1-3的整数,z为1,rm是选自由-ots、-oms、-bf3、-p-n(ch3)3-ph-,-p-no2-ph-、-sn(ch3)3-ph-、-i、-br和-cl组成的组中的离去基团,
更优选地,r’(rm)n表示在一个端甲基上被一个rm基团取代的甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、-(ch2o)x-ch3、-(ch2ch2o)y-ch2ch3或-(ch2ch2ch2o)z-ch2ch2ch3,其中x为1、2或3,y为1或2,z为1,rm选自由-ots、-oms、-otf、-br和-cl所组成的组。
根据具体实施方式,r’(rm)n可选自:-c(rm)3、-ch(rm)2、-ch2rm、-ch2c(rm)3、-ch2ch(rm)2、-ch2ch2rm、-ch2ch2c(rm)3、-ch2ch2ch(rm)2、-ch2ch2ch2rm、-ch(ch3)c(rm)3、-ch(ch3)ch(rm)2、-ch(ch3)ch2rm、-ch2ch2ch2c(rm)3、-ch2ch2ch2ch(rm)2、-ch2ch2ch2ch2rm、-c(ch3)2c(rm)3、-c(ch3)2ch(rm)2、-c(ch3)2ch2rm、-ch2och2rm、-ch2och2och2rm、-ch2och2och2och2rm、-ch2ch2och2ch2rm、-ch2ch2och2ch2och2ch2rm、-ch2ch2ch2och2ch2ch2rm,但不限于此。
rm最优选为ots。
根据具体实施方式,本发明优选的具体中间体为:
r’(rm)n=-ch2ch2och2ch2och2ch2ots的中间体化合物:
(2-(2-(2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-基)-1h-吲唑-1-基)乙氧基)乙氧基)乙基-4-甲基苯磺酸(2-(2-(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-1h-indazol-1-yl)ethoxy)ethoxy)ethyl4-methylbenzenesulfonate),和
(2-(2-(2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-基)-2h-吲唑-1-基)乙氧基)乙氧基)乙基-4-甲基苯磺酸)(2-(2-(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-2h-indazol-2-yl)ethoxy)ethoxy)ethyl4-methylbenzenesulfonate);
r’(rm)n=-ch2ch2och2ch2ots的中间体化合物:
2-(2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-基)-1h-吲唑-1-基)乙氧基)乙基-4-甲基苯磺酸酯(2-(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-1h-indazol-1-yl)ethoxy)ethyl-4-methylbenzenesulfonate),和
2-(2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-基)-2h-吲唑-1-基)乙氧基)乙基-4-甲基苯磺酸酯(2-(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-2h-indazol-1-yl)ethoxy)ethyl-4-methylbenzenesulfonate);
r’(rm)n=-ch2ch2ots的中间体化合物:
2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-及)-1h-吲唑-1-基)乙基-4-甲基苯磺酸酯(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-1h-indazol-1-yl)ethyl-4-methyl-benzenesulfonate),和
2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-及)-2h-吲唑-1-基)乙基-4-甲基苯磺酸酯(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-2h-indazol-1-yl)ethyl-4-methyl-benzenesulfonate)。
在一个尤其优选的实施方式中,所述中间体化合物为以下化合物1’或化合物2’:
本发明第三方面提供一种制备上述式i或式ii化合物的方法,所述方法包括使上述中间体与k18f或na18f进行一步法反应获得所述化合物的步骤。
根据一种实施方式,所述反应在40-150℃下进行5-60分钟。反应结束后冷却至约30℃。
所述中间体可事先溶于极性有机溶剂中,具体实例如dmf、dmso等。
根据优选实施方式所述的方法,进一步包括通过高效液相色谱或快速柱层析(例如,使用sep-pak的c18分离柱)进行纯化的步骤。洗脱液可为10%-100%的乙腈或乙醇,收集放射性组分。
本发明式i或式ii化合物的制备方法采用上述中间体通过一步取代直接制得,可使用全自动化合成设备(例如美国ge公司的tracelabfxcpro、fastlab2等)自动合成,从而减少或避免生产人员与放射性物质的接触。
本发明的方法合成效率高,标记率达50%-70%(未进行衰减矫正),比活度大于等于37gbq/μmol,放化纯大于95%,符合临床应用要求。
根据本发明的一个优选实施方式,该制备方法在取代反应之前,进一步包括通过以下步骤制备k18f或na18f:
步骤1,由回旋加速器轰击18氧-水生成18f-hf。
步骤2,使含18f-hf的靶水通过阴离子交换树脂柱(例如,qma小柱(waters,美国)),从而将18f-离子保留在树脂柱上,然后使用盐溶液从树脂柱将18f-洗脱下来,并使之进入反应瓶。
步骤3,向反应瓶中加入相转移催化剂和一定量的有机溶剂,通入氮气并进行加热,蒸干反应瓶中的溶剂,从而得到干燥无水的18f的氢氟酸盐。
根据本发明优选的实施方式中,步骤2中的所使用的盐溶液可为本领域常规使用的那些,例如可选自氯化钠、氯化钾、碳酸钾和碳酸氢钾中的一种或多种,相应地得到k18f或na18f。
在本发明另一优选的实施方式中,步骤3中所使用的相转移催化剂可为任何常用的催化剂,例如氨基聚醚(k2.2.2,可购自sigma-aldrich、百灵威、阿拉丁、tci等)。
所使用的有机溶剂并没有特别限制,例如可为乙腈、丙酮、乙醇中的一种或多种。
在又一具体实施方式中,步骤3中,可通过电热炉加热,也可采用微波加热。
根据一种实施方式,本发明的中间体可以gdc-0941(2-(1h-吲唑-4-基)-6-(4-甲基磺酰基-哌嗪-1-基甲基)-4-吗啉-4-基-噻吩[3,2-d]嘧啶),一种1型磷脂酰肌醇3激酶(pi3k)抑制剂为起始物来合成。
如下反应式所示,起始物gdc-0941(可商购)与rf-r’-(rm)n在有机溶剂中在碱的存在下生成单取代的中间体式i’/ii’混合物。其中所述有机溶剂例如可以是dmf、乙腈、dmso或丙酮等,但不限于此。所述碱可为无机碱,诸如碱金属氢氧化物(如,氢氧化钠)、碱金属碳酸盐(如,碳酸钠、碳酸钾、碳酸铯)碱金属氢化物(如,氢化钠、氢化钾);有机碱,诸如烷基胺(如,三乙胺、二异丙基乙胺)等。其中r’、rm、n如以上所定义;rf是选自由-ots、-oms、-otf,-i、-br和-cl组成的组中的离去基团;rm和rf可以相同,也可以不同。
如上反应式,以gdc-0941为起始物进行合成,通常获得吲唑基上两种n取代的混合物。
因此,本发明的方法包括获得包含相同的-r’(rm)n取代基的两种式i’和式ii’中间体混合物,不经分离直接通过上述一步法制备最终的具有相同-r基团的式i和式ii化合物的混合物。
所得最终含具有相同-r基团的式i和式ii化合物的混合物可不分离为单独的化合物,而直接以混合物形式进行应用,并不影响显像质量,因而简化了制备方法和步骤。
本发明的第四方面提供一种正电子显像剂,该显像剂包括至少一种上述式i和式ii所示的化合物。
根据一种具体的实施方式,所述正电子显像剂包括相同-r基团的式i和式ii化合物的混合物作为活性物质。
所述显像剂还可包括必要的溶剂和医用添加剂,例如生理盐水,少量乙醇等。
本发明的化合物是gdc-0941的衍生物。已知,gdc-0941(或称pictilisib)是新型选择性i类磷脂酰肌醇3-激酶(pi3k)抑制剂。gdc-0941竞争性地与pi3k的atp结合口袋相结合,阻止传递pi3k下游信号的第二信使磷脂酰肌醇3,4,5-三磷酸(pip3)的形成(folkesaj,ahmadik,aldertonwk,etal.theidentificationof2-(1h-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine(gdc-0941)asapotent,selective,orallybioavailableinhibitorofclassipi3kinaseforthetreatmentofcancer.jmedchem.2008;51:5522-5532;和knightza,shokatkm.chemicallytargetingthepi3kfamily.biochemsoctrans.2007;35:245-249)。
gdc-0941对pi3kα和pi3kδ亚型具有高选择性,而对pi3kβ和pi3kγ亚型选择性适中。在多个肿瘤细胞系中,包括a2780、mda-mb-361、pc3和u87mg,gdc-0941引起生长抑制(folkesaj,ahmadik,aldertonwk,etal.theidentificationof2-(1h-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine(gdc-0941)asapotent,selective,orallybioavailableinhibitorofclassipi3kinaseforthetreatmentofcancer.jmedchem.2008;51:5522-5532)。gdc-0941也可以抑制含有p110突变或pten缺失的trastuzumab敏感或抗性的her2扩增癌细胞的生长。在多个异种移植肿瘤模型中,gdc-0941也可以减少肿瘤体积(junttilatt,akitarw,parsonsk,fieldsc,lewisphillipsgd,friedmanls,sampathd,sliwkowskimx.ligand-independenther2/her3/pi3kcomplexisdisruptedbytrastuzumabandiseffectivelyinhibitedbythepi3kinhibitorgdc-0941.brjcancer.2011;104(7):1116-25)。
gdc-0941属于噻吩并[3,2-d]嘧啶类化合物。其依靠吗啉基-噻吩[3,2-d]嘧啶结构,特别是吗啉基结构与pi3k特异性结合(folkesaj,ahmadik,aldertonwk,etal.theidentificationof2-(1h-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine(gdc-0941)asapotent,selective,orallybioavailableinhibitorofclassipi3kinaseforthetreatmentofcancer.jmedchem.2008;51:5522-5532)。
由以上定义可知,本发明式i和式ii的化合物中携带放射性核素的烷基链或烷氧基/聚醚链远离有效结合部位(即,吗啉基-噻吩[3,2-d]嘧啶结构),对特异性结合没有特别影响,因而具有与gdc-0941相似的结合特性。因此,推测本发明的化合物也能够特异性与pi3k的atp结合口袋相结合。
经证实,当pi3k/akt/mtor信号通路被异常激活时,本发明的化合物能够在病灶处富集,从而产生放射性显影,有效指示pi3kpi3k/akt/mtor信号通路的高表达。
因此,本文的化合物和显像剂可应用于诊断pi3kpi3k/akt/mtor信号通路高表达的疾病,特别是肿瘤,如癌,例如其原发病灶(samuelsy,wangz,bardellia,etal:highfrequencyofmutationsofthepik3cageneinhumancancers.science,2004.304:554;yuantl,cantleylc:pi3kpathwayalterationsincancer:variationsonatheme.oncogene2008.27:5497-5510;ikenouet,kanaif,hikibay,etal:functionalanalysisofpik3cagenemutationsinhumancolorectalcancer.cancerres2005.65:4562-4567;samuelsy,velculescuve:oncogenicmutationsofpik3cainhumancancers.cellcycle2004.3:1221-1224)。所述肿瘤可以选自由乳腺癌、子宫颈癌、结肠癌、子宫内膜癌、胶质瘤、肺癌、黑色素瘤、卵巢癌、胰腺癌、前列腺癌、脑癌和霍奇金淋巴瘤所组成组中的一种或多种。所述肿瘤优选乳腺癌和结肠癌。
本发明所提供的靶向pi3k通路的化合物分子探针能够以高灵敏度、无创的pet显像实时地对受试者的pi3k的表达水平进行评估,有效地对pi3k高表达肿瘤进行鉴别诊断、分子分型、疗效评估、预后评价等。
具有18f的本发明的式i和式ii的化合物可具有约110分钟的半衰期。与常用的11c标记的放射性核素相比(其半衰期为约20分钟),能够实施更长时间的可视化监测,可进行较长时间的显像,具有更好的靶与非靶比值和更好的显像质量和对比度。
在本发明中制备式i和式ii化合物通过一步法合成,可利用全自动化合成仪自动合成,从而能降低操作人员射线辐射,并且反应快速,反应过程重复性高、产物稳定性好。产物放射产量高,根据优选实施例的化合物的非校正放射产量达70%或更高。根据优选实施方式获得的混合物,其标记率为50%-70%,比活度大于37gbq/μmol,放化纯大于95%。
附图说明
图1为实施例1中前体bar-2的hplc谱图,其中,19-20min中两个峰证实前体bar-2中存在互为同分异构体的两种化合物。
图2为实施例1中前体bar-2的1h-nmr谱图。
图3为实施例1中19f-bar-3标准品的hplc谱图,其中,10.5min和12.5min两个峰证实19f-bar-3中存在互为同分异构体的两种化合物。
图4为实施例2中18f-bar-3的放射性hplc谱图,其中,15min处的峰为18f-bar-3,无游离18f存在。
图5为根据实施例3,18f-bar-3和11c-bar-1在健康昆明鼠中的pet成像及代谢分析研究,其中,a示出了11c-bar-1的pet成像;b示出了18f-bar-3的pet成像;c示出了两者的肝脏摄取值比较。
图6为根据实施例4,18f-bar-3在pi3k高表达的mcf-7荷瘤鼠中pet成像结果(a)及生物分布代谢研究(b)。
图7为根据实施例4,11c-bar-1和18f-bar-3分别在mcf7荷瘤鼠和mda-mb-231荷瘤鼠中的静态显像结果,其中左边两图为18f-bar-3,右边两图为11c-bar-1,并且a均为mcf-7荷瘤鼠的静态显像结果;b均为mda-mb-231荷瘤鼠的静态显像结果。
具体实施方式
以下结合实施例和附图进一步描述本发明。应当理解,下列具体实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。
除非另有特殊说明,否则以下实施例中使用的原料和试剂均为市售商品,或者可以本领域公知方法来制备。
以下,将以尤其优选的实施例18f-bar-3来具体阐述本发明。
实施例1:19f-bar-3的制备
为了便于研究制备方法,先按以下反应式制备无放射性的化合物。
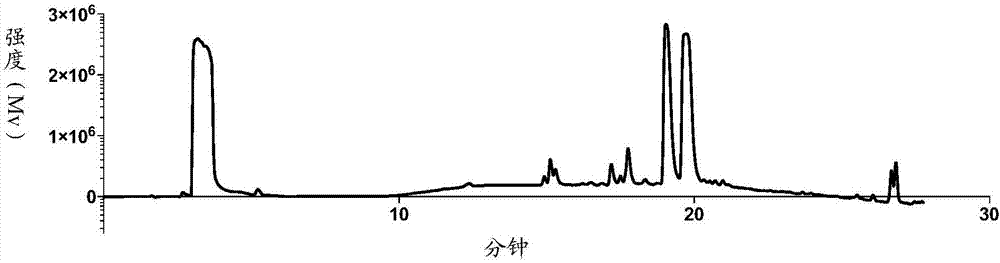
将0.5克bar-1(即gdc-0941,selleck,美国)与1.5克tso-peg3-ots(百灵威)溶解于50ml乙腈中,加入0.5克碳酸钾,于70℃下反应1~3天。待反应结束后,浓缩去除溶剂,残留物经硅胶柱层析纯化得产物0.4克,为淡黄色油状物,产率82%。所得产物经质谱(m/z(esi+),计算值799.25,实测值800.25,质谱仪expressioncms,advion)、1h-nmr(400mhzdrxnmrspectrometer,brukercorporation,参见图2)和hplc(岛津lc-20a,色谱柱菲罗门lunac18(2),250mm4.6mm,参见图1,其中,19-20min两个峰互为同分异构体)确认为下图所示bar-2同分异构混合物(2-(2-(2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-及)-1h-吲唑-1-基)乙氧基)乙氧基)乙基-4-甲基苯磺酸酯(2-(2-(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-1h-indazol-1-yl)ethoxy)ethoxy)ethyl4-methylbenzenesulfonate)和2-(2-(2-(4-(6-((4-(甲磺酰基)哌嗪-1-基)甲基)-4-吗啉噻唑[3,2-d]嘧啶-2-及)-2h-吲唑-2-基)乙氧基)乙氧基)乙基-4-甲基苯磺酸酯(2-(2-(2-(4-(6-((4-(methylsulfonyl)piperazin-1-yl)methyl)-4-morpholinothieno[3,2-d]pyrimidin-2-yl)-2h-indazol-2-yl)ethoxy)ethoxy)ethyl-methylbenzenesulfonate)。
随后,将所合成的2mgbar-2不需分离两种异构体直接与10mg四丁基氟化铵(tbaf)(二者摩尔比大于1:1)溶解在5mlthf中,室温下搅拌过夜。随后,通过旋转蒸发去除溶剂,残留物经硅胶柱层析纯化得产物0.5mg,淡黄色油状物,产率30%。所得产物经质谱(m/z(esi+):[m+h]+计算值647.2,实测值648.2)、hplc(参见图3,其中10.5min和12.5min峰互为同分异构体)确认为19f-bar-3(4-(2-(2-(2-(2-(2-氟代乙氧基)乙氧基)乙基)-2h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉和4-(2-(1-(2-(2-(2-氟代乙氧基)乙氧基)乙基)-1h-吲唑-4-基)-6-((4-(甲磺酰基)哌嗪-1-基)甲基)噻吩[3,2-d]嘧啶-4-基)吗啉)混合物。
实施例2:18f-bar-3的合成
本实施例采用美国ge公司的自动化合成设备(tracelabfxfn)进行全自动化一步法快速合成。
由回旋加速器(ge,minitrace)轰击3ml18氧-水(h218o)生成18f-hf500mci。使含18f-hf的靶水通过qma小柱(waters,accellplusqma)以将18f-离子保留在qma小柱上。然后用含3mg碳酸钾和15mg相转移催化剂k2.2.2(sigma-aldrich)的1.5ml乙腈水溶液(乙腈与水体积比2:1)将18f-从qma小柱上洗脱下来,并进入反应瓶,加热并通氮气,直至蒸干反应瓶中的溶剂以得到干燥无水的k18f(~450mci)。
将含2mg实施例1制备的bar-2的1.0ml无水dmso溶液加入到反应瓶中,于80℃下反应30分钟,然后冷却至30℃。随后用hplc(岛津lc-20a,色谱柱:菲罗门lunac18(2),250mm×10mm)进行分离纯化,并采用手动方法收集放射峰组分。用0.22μm的无菌滤膜(millipore,millex-fg)过滤,得到产物18f-bar-3混合物。
取18f-bar-310μci/20μl,通过放射性高效液相色谱(hplc:岛津lc-20a,色谱柱:菲罗门lunac18(2)250mm×4.6mm)测定放射化学纯度。起始500mci18f,终产物活度300mci±50mci(n=3),标记化合物18f-bar-3的标记率:60%±10%(n=3),放射性化学纯度大于95%。其放化纯测试结果如图4所示。
实施例3:18f-bar-3与11c-bar-1的生物分布与代谢的比较
通过pet显像考察本发明实施例2制备的18f-bar-3在正常昆明小鼠(雌性,4周龄,购自北京华阜康生物科技股份有限公司)体内的生物分布与代谢,并用11c-bar-1作为对照。
11c-bar-1的制备:
按下反应式采用美国ge公司的自动化合成设备(tracelabfxcpro)进行全自动化一步法快速合成11c-bar-1。
由回旋加速器(geminitrace)在外部生成500mci11co2。将500mci11co2传输进入反应器与100ml/minh2混合,在线生成11ch4500mci。随后与升华的碘(碘炉温度100℃,碘粒约20g)在720℃高温下反应生成碘甲烷(11ch3i,160mci)。
使300mci11ch3i进入自动化合成设备的反应瓶(室温,2mg化合物bar-1溶于5nnaoh/400μldmso中)。混合液65℃反应5min,然后冷却至30℃。
混合物进入hplc进行分离纯化,采用手动方法“peak-cut”收集放射峰组分。用0.22μm的无菌滤膜过滤,得到产物11c-bar-1(起始500mci碳-11,终产物约12.5mci±2.5mci(n=3),衰变校正产率:10.5%±2.5%,(n=3))。
取11c-bar-110μci/20μl,通过放射性高效液相色谱(hplc,岛津lc-20a,色谱柱:菲罗门lunac18(2),250mm×4.6mm)测定放射化学纯度。放射性化学纯度大于95%。
动态成像
使用raycantechnology有限公司的biocaliburnlh小动物成像系统进行动态成像。
首先使用氯胺酮/甲苯噻嗪(ketamine/xylazine,百灵威,110/10mg/kg)对正常昆明小鼠(每组3只)进行腹腔麻醉,将其俯卧位固定在micropet检查床上,然后采用床边尾静脉注射3.7mbq的18f-bar-3或11c-bar-1探针后,迅速放到扫描中心视野立即开始采集数据(6×10s,4×30s,4×60s,4×2min,5×5min共采集23帧),共采集40min左右。采用二维有序子集最大期望值法(osem,two-dimensionalorderedsubsetsexpectationmaximizationalgorithm)进行所有的图像后处理。
兴趣区(roi,regionsofinterest):在经衰减校正后的全身冠状位图像上通过手动勾画感兴趣的器官。定量分析各器官(roi)对放射性的摄取,得到相应部位每克组织注射剂量百分比%id/g。
通过对小鼠进行micropet动态成像能够更好地观察和比较18f-bar-3和11c-bar-1探针在体内的分布与代谢特征。参见图5,其中a示出了11c-bar-1的pet成像随时间的变化情况;图b示出了18f-bar-3的pet成像随时间的变化情况;c对比示出了两者的肝脏摄取值随时间变化的曲线。
从图5的a、b图可见,随着时间的推移18f-bar-3和11c-bar-1在肝脏的放射性浓聚均随时间逐渐降低,而胆、肠道部位放射性浓聚则逐渐升高,这表明两者均主要通过肝脏、胆、肠道排泄和代谢。
另外比较18f-bar-3(图5b)和11c-bar-1(图5a)的成像结果可以发现,与11c-bar-1相比,从5min开始,18f-bar-3成像的肝脏中与背景的对比度明显较低(参见图5b),这是因为18f-bar-3中引入了peg3作为连接基团改善了水溶性,使得其在肝脏中的代谢加快。从图5c中的定量的结果看,18f-bar-3在肝脏中的总清除速率明显比11c-bar-1快,虽然一开始前者肝脏摄取更高,但是从5分钟到35分钟的18f-bar-3在肝脏的摄取明显低于11c-bar-1。
此外,peg3连接基团的引入还增加了探针在血液中的滞留时间,在第30min时,18f-bar-3血液中摄取显著高于11c-bar-1(3.19±1.05%id/gvs1.34±0.66%id/g),因而有利于探针在肿瘤等靶部位的蓄积。
由图5可见,本发明的半衰期为110min的18f标记的探针,可以进行更长时间的显像,并且具有更好的在体显像结果并且,对比实施例2对本发明化合物的制备以及本实施例对11c-bar-1的制备,本发明的化合物18f标记的探针的产率高(50-70%)。11c标记步骤较18f标记更为复杂,需要多步工序才能从加速器制备的11c-co2转化为11c-bar-1标记所需的放射性前体11c-ch3i,而18f标记只需要将加速器制备的18f通过简单的离子键换树脂处理即可用,因此18f-bar-3制备工序整体比11c-bar-1更少且更简单、标记可控且稳定。18f-bar-3一次生产可供10人以上病人使用(按每人0.1mci/kg计算),有利于探针的临床广泛使用;相比之下,11c标记的探针产率有限(8-13%),一次生产仅可供1~3人使用。
实施例4:18f-bar-3在荷瘤小鼠体内的生物学性质
通过pet显像考察本发明实施例2制备的18f-bar-3在荷瘤小鼠模型体内生物学性质。
动物肿瘤模型构建:balb/c-nu裸鼠(雌性、4周龄)由北京华阜康生物科技有限公司提供,在华中科技大学动物实验中心无特殊病原体屏障系统饲养。实验中用到的所有动物都经过华中科技大学同济医学院实验动物使用和管理委员会审核。使pi3k低表达mda-mb-231细胞(中国科学院典型培养物保藏委员会细胞库)、pi3k高表达的mcf-7细胞(中国科学院典型培养物保藏委员会细胞库)各5×106个分别悬浮于100μlpbs中,并与等体积的人工基底膜(matrigel,美国bd)混匀,皮下种植在balb/c-nu裸鼠右上肢肩背部,待肿瘤直径为0.7cm~1.0cm时进行动物实验研究。
动态成像:首先使用氯胺酮/甲苯噻嗪(百灵威,110/10mg/kg)腹腔麻醉模型构建成功的mda-mb-231、mcf-7荷瘤鼠,并将其俯卧位固定在micropet检查床上,然后采用床边尾静脉将大约3.7-5.55mbq(100-150μci)的18f-bar-3注入体内后,立即放到扫描中心视野开始采集数据(6×10s,4×30s,4×60s,4×2min,3×5min,3×10min共采集24帧),共采集60min。静态成像在显像剂注射1h后用实施例3中的小动物pet成像系统采集数据10min。实验中使用进行腹腔麻醉。采用二维有序子集最大期望值法(osem)进行所有的图像后处理。
兴趣区(roi):在经衰减校正后的全身冠状位图像上通过手动勾画感兴趣的器官或肿瘤。定量分析肿瘤部位和其他器官对放射性的摄取,得到相应部位每克组织注射剂量百分比%id/g。对于肿瘤/肌肉比值的计数,则首先roi勾画肿瘤部位感兴趣区,然后再勾选对侧肌肉为背景。
图6示出了18f-bar-3作为分子探针在mcf-7荷瘤鼠中的成像结果(a)及生物分布代谢研究(b)。从图6a中可以看出,与正常鼠相同,肝脏作为最主要的排泄途径其相应部位放射性浓聚随时间逐渐降低,而在mcf-7肿瘤在注射探针10s后便清晰可见,在所测试的30min-2h时间区间mcf-7肿瘤具有很好的对比度。探针在mcf-7肿瘤中的放射性浓聚随着时间的延长逐渐增加,在1h时最高,在此后的1h中缓慢降低,并仍保持较高的摄取。从图6b中的定量结果可以看出,18f-bar-3在mcf-7肿瘤中的摄取在十分钟左右变到达最大值,并在所测试的2h内保持在6~7%id/g的较高水平。与此同时,肌肉的摄取值较低,只有~1%id/g。肿瘤与肌肉的摄取比值为6-7。
图7示出了mcf-7荷瘤小鼠和mda-mb-231荷瘤小鼠分别注射18f-bar-3与11c-bar-1后1h的pet显像结果。左边两图为18f-bar-3的显像结果,右边两图为11c-bar-1的显像结果。a均为mcf-7模型鼠在60min时的静态显像结果,b均为mda-mb-231模型鼠在60min时的静态显像结果。
由图7可见,18f-bar-3和11c-bar-1在pi3k高表达肿瘤mcf-7中的蓄积显著高于在pi3k低表达肿瘤mda-mb-231的蓄积。显示18f-bar-3和11c-bar-1在肿瘤中的摄取与pi3k的表达量有一定的相关性,因而证实了18f-bar-3和11c-bar-1能够特异性提示pi3k的异常高表达,并进而用于筛选出pi3k蛋白抑制剂敏感病人或者对pi3k/akt/mtor信号通路抑制剂类药物的疗效提供评估。
从图7中可以进一步看出,与11c-bar-1相比,18f-bar-3在小鼠内的整体摄取值增加,这与所增加的peg3连接基团有助于降低血液清除速率有关。
注射两个探针1h后,mcf-7肿瘤均清晰可见,具有较好的对比度,但是18f-bar-3的摄取绝对值显著高于11c-bar-1,且18f-bar-3在肝脏等腹部脏器中的放射性浓聚显著低于11c-bar-1。而且18f-bar-3肿瘤摄取值和肿瘤与背景比值均比11c-bar-1高。对于pi3k表达相对较低的mda-mb-231荷瘤小鼠模型,在注射11c-bar-1探针后,肿瘤部位未见特异性摄取,而在肝脏等腹部脏器种放射性浓聚较高,这可能与11c-bar-1在血液中清除较快有关。而注射了18f-bar-3后1h,mda-mb-231肿瘤清晰可见,而且在肝脏中未见明显的放射性浓聚,仅在胆囊、膀胱和小肠中有较高的放射性浓聚。这表明经peg3修饰的18f-bar-3探针具有比11c-bar-1更好的药代动力学属性,且同时保持甚至增加了与pi3k高表达肿瘤的亲和力。
- 还没有人留言评论。精彩留言会获得点赞!