乙肝病毒肝内整合状况的感染者外周血检测方法与流程
本发明涉及乙肝病毒整合状况的检测,特别涉及外周血游离dna在乙肝病毒整合状况检测中的应用。
背景技术
乙肝病毒(hepatitisbvirus,hbv)作为逆转录病毒,具有整合入宿主基因组的能力,最早在20世纪八十年代早期就被报道,在肝癌基因组中的研究也有很多积累1-13。hbv的整合能力并不是其生活史(lifecycle)以及感染宿主所必须,肝病毒基因组中目前不认为存在基因编码有整合酶活性的病毒蛋白14,早期的研究曾认为hbv整合事件是在全基因组随机分布的罕见事件15。hbv整合机制的研究认为,病毒整合入宿主基因时的主要形式是线性双链dna(doublestrandlineardna,dsldna)16,是cccdna形成过程中的中间体,而cccdna是hbv感染慢性化中最关键的一环17,因而推断整合事件可以伴随着hbv慢性感染的全过程。虽然研究人员不断尝试构建可以用于hbv生活史研究的细胞、动物模型,但一直以来尚未有成熟体系18,对cccdna相关的生物学过程研究基于鸭乙型肝炎病毒(dhbv),包括目前估计的103~104个感染的细胞中会发生一次整合也是基于dhbv感染实验16。最早研究者曾认为整合入宿主基因组中的病毒片段处于失活(defective)状态无法产生蛋白,后续研究提示整合频率最高的hbv基因组片段为dr2-dr1区域以及pres/s基因区域19,会分别翻译产生截短型的x蛋白、表面蛋白20,并通过各自特定机制诱发肝细胞向肿瘤细胞转变21,22,因而此类现象也被认为属于整合事件在肝脏病程进展中的反式(trans)作用。最新研究报道,整合的片段与邻近的基因可产生嵌合转录本(chimerictranscripts),如hbv-mll423、hbv-cyclina224;chiuyt等人提出存在pre-mrnasplicing机制,使其可以翻译出无法被宿主细胞降解的病毒-人嵌合蛋白,导致细胞周期进程加快24。近年来更是认为慢性感染中整合的病毒dna片段也会继续在受累的人肝脏细胞中翻译产生病毒的蛋白质影响一系列细胞的功能活动以及宿主的免疫反应25,26。
hbv整合除了在不同感染者间存在一定异质性(heterogeneity)外,在同一患者体内也可以观测到多次的整合事件。2012年,sungwk等人在对81例hbv感染后肝癌的wgs结果中发现近400个整合事件,单个肿瘤最多存在16个不同的hbv整合,其中年轻肝癌患者肿瘤组织中可以观测到更多的病毒整合19。此外,2015年yanh等人的研究也发现在病毒感染后早发肝癌(<30岁)患者中的整合模式与晚发肝癌(>70岁)患者中存在差别,在前者肝脏肿瘤组织中发现了更多可能影响肝细胞的病毒整合事件27。因而,上述发现亦支持hbv整合可能赋予肝细胞生长优势的观点,整合可以加速肝脏病变发展的进程。同时,肿瘤的优势克隆中可均含有同一特定hbv整合,基于此miaor等人通过肿瘤全基因组测序数据分析hbv整合在2例肝内多发肿瘤中的差异,成功鉴别出肿瘤的克隆起源:具备相同hbv整合的多肿瘤病灶属于单克隆起源,hbv整合事件不同则为多克隆起源,两者可具有不同的临床预后28。2008年流调查数据显示,中国约有慢性hbv携带者9300万人,其中慢性乙型肝炎患者1900万人,虽然乙肝疫苗的广泛应用使得感染得到了非常有效的控制,但慢性感染人群仍具有相当大的基数,其肝脏病变发展的评估和肝癌的预警一直是病毒性肝炎治疗中的重点。
上世纪八十年代肝癌中的整合病毒片段分析多基于限制性酶切或者southern杂交技术,获得的目标片段通过一代测序进行后续验证确定1-9。实验流程繁琐、分析通量低,因而无法从全局上分析整合事件的生物学影响。1995年,minami等人开发了alupcr策略在基因组中筛查整合位点使得病毒整合分析的通量得以提升29,30。然而该策略的前提是病毒整合需要发生在alu片段附近,这种前提假设破坏了分析的无偏性,不处于alu片段区域的整合是无法被该技术捕捉到的。此外,pcr技术对片段的扩增中,会因为其固有的templateswitching的问题造成假阳性的结果,即把肝癌组基因组和游离病毒dna在扩增时连接在一起31,32。
近几年随着高通量测序技术的广泛应用,研究人员在大量肝癌样本中开展全基因组序列测定(wholegenomesequencing,wgs)或基于alu-pcr对整合位点的筛查19,其中2014年lix等人通过荟萃已报道的1115次整合事件,对hbv在肝癌基因组中的发生频率、整合规律有了更深入的刻画33。目前认为,整合事件多发生在染色体的转录活跃的区域,可能通过顺式(cis)调控作用影响临近基因的表达17。虽然这种表达活性的变化在肿瘤发生发展中的作用仍需深入探讨,但存在高频整合靶向基因被多个研究共同报道,并且多涉及细胞的存活、增殖和永生化等生物学过程,例如端粒酶逆转录酶基因(tert)、组蛋白甲基转移酶(mll4)以及细胞周期蛋白e1(ccne1)等19,28,34。此外,整合发生的区域也存在典型的序列特征,更容易出现在基因组不稳定的区域如重复序列、长散在重复序列、短散在重复序列、微卫星、端粒、中心粒区域35,36,甚至整合位点本身存在基因组的结构变异37,一些染色体(5,、16、17、19号)被确定更容易发生hbv病毒整合38-43。然而全基因测序技术的成本依旧十分昂贵,尤为重要的是,80-90%hbv感染后肝癌组织中可发现病毒的整合事件,因而其在肝癌发生发展中的作用不容忽视44。
液体活检(liquidbiopsy)技术近年来迅速发展,在外周血中检测游离dna(cellfreedna,cfdna)成为监测患者疾病病程进展的重要手段45。hbv感染造成的肝脏损伤程度不同时,患者外周血中cfdna水平存在差别,例如在hbv感染后急性肝衰竭患者中,cfdna水平的升高可帮助判断患者的预后46;除了cfdna数量的变化,一些慢性携带者的cfdna上可以检测到tp53基因的突变,提示此类患者肝脏肿瘤发生风险可能会提高47;已有尝试通过监测hbv感染不同阶段血浆cfdna的甲基化改变,发现cfdna中nf300、slc22a20和shisa等基因的甲基化水平在慢肝、肝硬化和肝癌患者中存在差异,将其作为筛选肿瘤高风险人群的标志物48。然而,利用cfdna检测突变和甲基化等情况所能提供的信息仍然十分有限,有必要探索提供更多信息量的手段。病毒整合作为hbv感染后肝细胞基因组中的特征性事件,目前国内外尚未有研究探索整合位点的dna片段是否能被释放至外周血,更无研究探索其是否有足够的特异性和灵敏性以用于检测分析。
技术实现要素:
本发明的发明人首次发现hbv整合位点的dna片段能够被释放至外周血,并且能够用于检测分析。本发明的一个目的在于提供一种检测受试者的乙肝病毒整合状况的方法,可通过液体活检等非侵入方式实现,以避免手术活检和穿刺活检等组织活检方式所存在的安全性风险以及采样困难、样品异质性等缺陷。
为了解决上述技术问题,本发明提供了一种检测受试者的乙肝病毒整合状况的方法,该方法以外周血中的游离dna作为样品进行检测。
作为优选,所述乙肝病毒整合状况是受试者肝组织或肝细胞的乙肝病毒整合状况,通过检测外周血游离dna中的乙肝病毒整合事件,以反映出受试者肝组织或肝细胞的乙肝病毒整合状况;优选地,所述乙肝病毒整合状况是肝组织或肝细胞中的乙肝病毒整合事件谱。
作为优选,所述检测受试者的乙肝病毒整合状况的方法包括如下步骤:(1)从受试者获取外周血样品;(2)检测外周血游离dna中的乙肝病毒整合事件。
在一些实施方式中,外周血游离dna中乙肝病毒整合事件的检测包括如下步骤:
(a)提取外周血游离dna;
(b)对步骤(a)提取的外周血游离dna进行末端修复,与接头连接,纯化连接产物;
(c)任选地,对步骤(b)获得的dna进行预扩增,纯化扩增产物;
(d)使用能够与乙肝病毒基因组序列杂交的探针在步骤(b)或步骤(c)获得的dna产物中进行捕获;
(e)对步骤(d)获得的捕获产物进行测序;
(f)通过生物信息学分析确定乙肝病毒整合事件。
在另一些实施方式中,外周血游离dna中乙肝病毒整合事件的检测包括如下步骤:
(a)提取外周血游离dna;
(b)对步骤(a)提取的外周血游离dna进行单分子测序或纳米孔测序;
(c)通过生物信息学分析确定乙肝病毒整合事件。
本发明的另一个目的在于提供一种检测受试者cfdna中乙肝病毒整合事件的方法。为了解决这一技术问题,本发明提供如下技术方案:
一种检测受试者cfdna中乙肝病毒整合事件的方法,所述包括如下步骤:
(a)提取外周血游离dna;
(b)对步骤(a)提取的外周血游离dna进行末端修复,与接头连接,纯化连接产物;
(c)任选地,对步骤(b)获得的dna进行预扩增,纯化扩增产物;
(d)使用能够与乙肝病毒基因组序列杂交的探针在步骤(b)或步骤(c)获得的dna产物中进行捕获;
(e)对步骤(d)获得的捕获产物进行测序;
(f)通过生物信息学分析确定乙肝病毒整合事件。
一种检测受试者cfdna中乙肝病毒整合事件的方法,所述包括如下步骤:
(a)提取外周血游离dna;
(b)对步骤(a)提取的外周血游离dna进行单分子测序或纳米孔测序;
(c)通过生物信息学分析确定乙肝病毒整合事件。
作为优选,步骤(a)中,提取外周血游离dna后,测定游离dna的片段分布,如果片段分布不符合150-200bp的理想范围,打断片段以使其符合理想范围。
作为优选,步骤(b)中,所述末端修复是在提取的外周血游离dna中加入末端修复缓冲液和末端修复加a酶混合物,进行末端修复反应。
作为优选,步骤(b)中,所述与接头连接在末端修复产物中加入接头和连接试剂进行连接反应。
作为优选,步骤(b)中,所述纯化连接产物是使用核酸纯化微珠对连接产物进行纯化。
作为优选,步骤(c)中,所述预扩增是对步骤(b)获得的dna进行pcr扩增;任选地,纯化扩展产物后进行干燥。
作为优选,步骤(c)中,所述纯化扩增产物是使用核酸纯化微珠对扩增产物进行纯化;任选地,在纯化后的扩增产物中加入能够阻断或妨碍接头序列间配对结合的寡核苷酸,和/或能够与人类基因组上的重复序列结合的寡核苷酸。
作为优选,步骤(d)中,在磁珠上进行捕获反应;优选地,所述磁珠为链霉素亲和磁珠。
作为优选,步骤(d)中,捕获后进行pcr扩增和纯化。
作为优选,步骤(e)中,测序量为1g、1.5g或2g;优选2g。
测序数据中的乙肝病毒整合事件虽然可以通过常规的生物信息学分析手段识别,但为了提供更好的识别效果,本发明提供了创新的生物信息处理方法,包括如下步骤:
(a)对测序读长进行比对,识别来源于受试者基因组的序列以及来源于乙肝病毒的序列;
(b)移除冗余读长;
(c)根据来源于受试者基因组的序列和来源于乙肝病毒的序列,找出受试者-hbv嵌合读长并确定其断点;
(d)根据两端断点确定整合事件。
作为优选,在判定整合事件之前,还包括对外周血游离dna的测序数据进行筛选的步骤。作为优选,具体包括:(i)滤除低质量测序数据;和/或(ii)滤除接头污染序列。
本发明进一步提供一种检测受试者肝细胞过量增殖的方法,所述方法包括如下步骤:
(1)采用本发明所述的方法检测受试者的乙肝病毒整合状况;或者采用本发明所述的方法检测受试者cfdna中乙肝病毒整合事件;
(2)在步骤(1)的检测结果中寻找指示肝细胞过量增殖的整合事件,所述指示肝细胞过量增殖的整合事件的识别标准为:整合事件中来源于受试者基因组的序列与其相应参比序列之间的核苷酸差异不超过2个,来源于乙肝病毒的序列与其相应参比序列之间的核苷酸差异不超过5个,并且同一整合事件每一端的断点都在至少2个非冗余的跨断点读长中存在;
(3)受试者存在符合所述识别标准的整合事件,则其肝细胞存在过量增殖。
本发明进一步提供一种肝癌的诊断/筛查方法,所述方法包括如下步骤:
(1)采用本发明所述的方法检测受试者肝细胞的过量增殖;
(2)对存在肝细胞过量增殖的受试者进行肝癌影像学检测。
本发明进一步提供外周血游离dna在检测或监测受试者乙肝病毒整合状况中的应用。
本发明进一步提供外周血游离dna在检测受试者乙肝病毒整合事件中的应用。
本发明进一步提供外周血游离dna在检测受试者肝细胞过量增殖中的应用。
本发明进一步提供外周血游离dna在诊断或筛查肝癌中的应用。
本发明进一步提供游离dna制备试剂在制备用于检测受试者的乙肝病毒整合事件的试剂和/或试剂盒中的用途。
本发明进一步提供游离dna制备试剂在制备用于检测受试者的乙肝病毒整合状况的试剂和/或试剂盒中的用途。
本发明进一步提供游离dna制备试剂在制备用于检测受试者肝细胞过量增殖的试剂和/或试剂盒中的用途。
本发明进一步提供游离dna制备试剂在制备肝癌诊断试剂和/或试剂盒中的用途。
本发明的方法可以在诊断或筛查过程中应用,也可以用于非诊断目的。
本发明所称的“整合”是指来自病毒的dna插入宿主的基因组中,所述来自病毒的dna可以是病毒基因的片段、病毒基因组的大部分区域或者整个病毒基因组。
本发明所称的“断点”是指病毒基因组或基因片段插入宿主基因组后病毒序列和宿主序列的交界点;
本发明所称的“受试者-hbv嵌合读长”、“跨断点读长”是指一个测序读长的序列同时覆盖断点周围的病毒基因序列和宿主基因序列;
本发明所称的“受试者”是哺乳动物,优选人类;所述人类受试者优选肝癌患者、乙肝患者、乙肝病毒携带者或者正常人。
本发明所称的“参比序列”是指的公共数据库中提供的已经确定碱基序列组成的基因组信息,用在基因组重测序中的测序读长比对,确定测序读长覆盖的基因序列在该基因组上的位置。受试者基因组和乙肝病毒的参比序列(或称参考序列)可以从现有技术文献或者在线序列数据库中获得,例如美国国立生物技术信息中心网站(https://www.ncbi.nlm.nih.gov/)。
本发明与现有技术相比具有如下优点和有益效果:本发明的方法对患者基本无创伤,可取代传统组织活检。以外周血中的游离dna作为样品来检测和监测受试者的乙肝病毒整合状况,相比于现有技术的常规做法,即通过手术活检或穿刺活检取组织样品后进行测试,能够避免并发症等风险事件,提高安全性,可以实现更高频繁的定期监测,而且避免了组织取样的异质性偏差。而且本发明的方法特异性好、灵敏度高,目前数据表明可以在100%的hbv感染后肝癌患者的外周血样本中检测到,最低可以在5ml外周血血浆提取到的全部cfdna中检测到片段。
附图说明
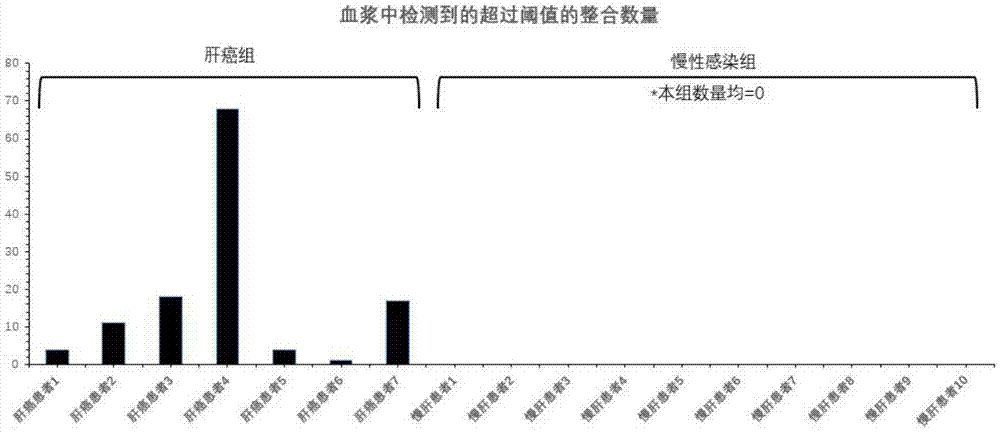
图1是肝癌组与慢性乙肝组cfdna中整合事件水平的比较图。
图2是肝癌组cfdna中整合事件与肝癌组织中整合事件的丰度比较图。
图3是最佳筛选标准下cfdna和肝癌组织中断点丰度的比较图。
图4是肝癌组与慢性乙肝组cfdna的片段插入/断裂模式图。
具体实施方式
本发明中,除非特别指明,所使用的试剂、设备等均为本领域常用的,或者可以通过市场购买获得。下面结合具体实施例详细描述本发明,应当理解的是这些实施例仅用于例证本发明,本发明并不仅局限于这些实施例,本领域技术人员根据本发明的揭示,在本发明的精神和原则之内所作的任何修改、等同替换、改进等,均包含在本发明的保护范围之内。
实施例1、样品采集和外周血游离dna的提取
在北京佑安医院收集了7例hbv感染后肝癌患者的外周血样。对于每个肝癌患者,除在手术前收集了外周血样本外,还在手术后收集了肝癌组织样本2份和癌旁肝组织样本2份,用于对照外周血中的病毒整合事件的发现能力。上述患者包括女性3人,男性4人,平均年龄51岁。
提取了外周血样本的cfdna以及肝癌组织样本和癌旁肝组织样本的dna。dna的提取采用qiagendneasyblood&tissuekits试剂盒,按照试剂盒的标准化流程提取。cfdna的提取采用以下步骤,并进行定量和质量评估:
1)使用游离dna采血管(streckcell-freedna
2)离心分离上层血浆,离心条件为:4℃、3000×g、15分钟。5ml血浆利用游离核酸提取试剂盒qiaampcirculatingnucleicacidkit(qiagen,valencia,ca)提取外周血游离dna,洗脱阶段使用20ul无dnase酶的超纯水。
3)荧光定量仪(
4)使用agilent2100生物芯片分析系统仪(2100bioanalyzerinstrument,dna1000assay)分析cfdna的片段分布。如果片段分布不符合150-200bp的理想范围,酌情采用超声(covarisfocused-ultrasonicators,covaris,inc.ma)打断片段,以使其符合理想范围。打断后的产物重复实施步骤3)-4),分析评估样本浓度及片段大小是否达到后续实验反应的要求。
结果显示,多数患者样品所提取的cfdna具有集中在167bp峰值的短片段分布,总量约600ng,全部用于后续的捕获实验。
实施例2、外周血游离dna的末端修复、接头连接和预扩增
对实施例1获得的cfdna按照如下步骤进行处理:
1)将提取获得的cfdna用超纯水定量至50ul体积,加入7ul末端修复缓冲液(endrepair&a-tailingbuffer)以及3ul末端修复加a酶混合物(endrepair&a-tailingenzymemix),混合后至60ul,在热循环仪上完成末端修复反应,反应条件为:20℃30分钟,65℃30分钟,4℃保存,热盖保持在70℃。
2)将上述60ul产物加入5ul的接头(接头包含后续扩增用引物区域、个体标签识别序列,与后续测序平台所需的接头对应),在冰上进行连接反应:加入55ul的连接反应混合液,包括5ul无核酸酶水,30ul的连接缓冲液,10uldna连接酶。20℃孵育15分钟(无热盖)。对于dna量比较少的样本,为了提高连接效率可以将孵育时间延长至不超过4小时,或者4℃过夜。
3)取110ul的连接产物,加入88ul平衡至室温的核酸纯化微珠(agencourtampurexpbeads),轻柔混合5-10次,并避免气泡产生,室温孵育5分钟。将离心管放至磁力架上,1分钟内磁珠成团,上清清亮,移除上清;在磁力架上的管中继续加入200ul80%乙醇,室温孵育30秒后吸除;重复2次;室温干燥磁珠3-5分钟,避免过分干燥造成产物产量下降。取22ul无核酸酶水洗脱dna。
4)20ul洗脱的dna,加入高保真dna聚合酶预混液25ul,加入扩增引物各2.5ul配制成50ul反应体系,所述扩增引物对应于接头中的扩增用引物区域,进行pcr扩增构建1ug的测序文库。
5)产物用核酸纯化微珠(agencourtampurexpbeads)进行前述纯化,用30ul无核酸酶水溶解纯化后的dna,获得产物约28ul。通过agilent2100生物芯片分析系统(2100bioanalyzerinstrument,dna1000assay)分析了片段长度,结果显示约为275-325bp左右。
6)测定前一步骤所获得的纯化产物的浓度,取一定体积的溶液,使得其中含有750ng纯化产物,加入1ul阻断液1、5ul阻断液2和5ul阻断液3,在冻干机上转换为干粉。其中,阻断液1和2中分别含有寡核苷酸block1和block2,block1和block2被设计用于阻断接头序列间的配对结合导致的捕获效率下降,阻断液3中含有寡核苷酸block3,block3被设计为结合人类基因组上的重复序列等以避免其对实验的干扰。
实施例3、杂交捕获实验
对外周血cfdna以及肝癌组织和癌旁组织dna进行整合事件的捕获,按如下步骤进行:
1)病毒整合捕获探针组的设计:根据ncbi数据库中整理获得的乙肝病毒a-h型的序列,包括中国地区流行的b型212条,c型457条,设计覆盖病毒全基因组3.2k的简并探针,每个探针长度约为100bp。探针的设计方法是本领域公知的,采用常规设计方法,例如常见的软件或网站即可完成,本实施例中的探针组具体是通过常用的引物设计网站https://design.igenetech.com/设计获得。
2)准备反应溶液:取实施例2获得的外周血cfdna干粉样品,加入无核酸酶水定量至10ul,轻柔混匀,转移入新的500ulpcr管(b管)内,置于冰上;200ulpcr管(a管)内加入20ul杂交缓冲液,预热至65℃;500ulpcr管(c管)内加入5ul核糖核酸酶阻断液(rnaseblock)以及2ul探针置于冰上,其中探针浓度根据cfdna产物的量进行了相应调整。
实施例1中制备的肝癌组织dna和癌旁组织dna也采用同样方法处理。
3)捕获反应体系配制:热循环仪器设定程序95℃5分钟,65℃保持,热盖100℃。之后放入b管启动程序。当仪器显示65℃时,将a管在热循环仪中放置5分钟,之后放入c管保持2分钟;从a管取13ul杂交缓冲液放入c管,并把b管的10ul样本也加入c管,避免过程中产生气泡。严格密封好c管后,在热循环仪上65℃孵育16-24小时,热盖100℃。
4)链霉素亲和磁珠准备:dynabeadsmyonestreptavidint1磁珠50ul放入500ulpcr管中,将管子置入磁力板(dynamag-96side),磁珠成团后移除上清,将管子从架子上取下,加入200ul结合缓冲液,轻柔混匀;重新将管子放置在架子上,磁珠成团,移除上清,注意勿打散磁珠,重复上述清洗步骤3次,之后用200ul结合缓冲液重悬磁珠立即用于下面的捕获反应。
5)链霉素亲和磁珠的捕获反应:将上述200ul磁珠直接加入热循环仪65℃中的500ulpcr管c,轻柔混匀;室温将混合物在旋转混匀器上10rpm混合30分钟;之后放在磁力板dynamag-96side上,磁珠成团后弃上清,加入200ul洗脱液1(常温低盐液洗脱液),取下磁力板后在旋转混匀器上室温10rpm混合15分钟。震荡管子后放回磁力板,磁珠成团后弃上清;加入200ul洗脱液2(高温高盐洗脱液),混匀后在混合器(thermomixer)上65℃孵育10分钟,速度为800rpm;震荡管壁后放入磁力板,磁珠成团后弃上清,重复洗脱液2的此步骤3次,之后确保其完全弃除干净。然后用30ul无核酸酶水重悬磁珠。
6)捕获后pcr及纯化:30ul上一步产物加入缓冲液18ul、对应于接头中扩增区域的通用扩增引物(此引物含有标签序列用于标记区分不同文库)各1ul、dna高保真酶1ul配成50ul的反应体系,混合后在热循环仪上pcr扩增,pcr条件为:热盖100℃;95℃4分钟后进入循环,循环过程为98℃20秒、60℃30秒、72℃30秒,共10个循环;之后72℃5分钟,12℃保持。然后把平衡至室温的核酸纯化微珠(agencourtampurexpbeads)(至少室温30分钟)55ul加入到管中,吹打5-10次(注意避免产生气泡),室温孵育5min。在磁力板上磁珠在1分钟内结块,弃上清,用200ul80%乙醇室温孵育30秒后移除,反复2次。室温下让磁珠干燥,从磁力板取下管子,用25ul无核酸酶水洗脱。之后放回磁力板,2分钟磁珠充分成团后将获得的23ul产物移到新的管中。使用agilent2100生物芯片分析系统(2100bioanalyzerinstrument,dna1000assay)分析了片段长度,结果显示约为275-325bp左右。
实施例4、捕获产物的测序
为了确定捕获整合事件所需的最佳测序量,构建了基于hepg2.2.15的阳性参照系统。hepg2.2.15细胞系作为hbv研究中最为著名的细胞系,其中整合有完整的hbv全长基因组序列,具有明确的整合位点。培养的hepg2.2.15在收获后通过乙肝病毒core抗体移除分泌产生的病毒颗粒,利用细胞核分离试剂盒纯化得到其细胞核,以排除细胞质中的cccdna对于标准定量的影响,采用过量探针合成的策略,保证捕获的敏感性。分别采用1g、1.5g、2g的测序量进行测序,使用的测序平台为illuminax-ten测序仪,测序步骤采用测序平台的标准化方法。
结果如下表显示,测试探针设计了三次实验,分别设定了1g测序量、1.5g测序量、2g测序量,捕获结果显示,在针对hepg2.2.15中的5个整合断点中,可以稳定发现染色体1、7、9上的断点,但是对于染色体2上的断点通过1g测序量不能发现,在测序量提升到2g测序量时可以发现。
事实上,最佳测序量可以随着所使用的探针组合的变化而调整,考虑到实际样本间可能存在的对测序深度要求的差别,例如cfdna量少、来自hbv的序列有限、或者病人本身释放入血的hbv整合片段不够充足等情况,为了尽可能的保证准确性和灵敏度,最终选择了较大的测序量进行后续试验,以免部分样本的测序深度不够导致无法发现整合事件。
根据以上结果,对实施例3中获得的经纯化的捕获产物进行高通量测序文库构建,采用illuminax-ten平台的商业化建库流程,然后进行了测序,每个文库测定约2g数据量。测序量为2g时,结果显示测序结果符合以下质量要求:rawbases不低于2000m碱基,质控后测序量(cleanbases)不低于1600mbases(qcrate>80%),平均片段读长不小于120bp,读长冗余率(duplicationrate)不大于1%,对于hbv基因组整体的覆盖度,4x的区域不低于90%,50x的区域不低于50%。
实施例5、生物信息学分析
对实施例4获得的测序结果进行生物信息学分析。首先用cutdapt软件等移除测序中的低质量及残余的接头序列,通过序列比对软件bwa将质控后测序读长比对到人及hbv参考基因组上,确定对于hbv基因组的覆盖度,通过picard软件移除比对后的冗余读长,之后评估测序的有效数据量及质量。调出数据中人-hbv嵌合读长(junctionreads)亦即覆盖断点的测序读长,分别对hbv端和人端进行基因组的序列注释,从而确定整合事件的发生特征:病毒整合的区域、人基因组上的位置等。
根据本领域的公知常识,cfdna大多来源于正常组织而非肿瘤,通常认为肿瘤患者的cfdna中循环肿瘤dna(circulatingtumordna,ctdna)的比例约为0.1%-1%之间,基于这一认识可以预期,cfdna中来自非癌肝组织的整合事件应当多于来自肝癌组织的整合事件。然而在对7例肝癌患者的外周血、肝癌组织和癌旁组织的整合事件进行比较和分析后(结果如下面的表格所示),出乎意料地发现,外周血cfdna中捕获到的23个整合事件全都能在肝癌组织中捕获到,但在正常肝组织中只能捕获到其中的18个。癌细胞的数量远少于正常的肝组织,癌细胞发生死亡裂解的频率也低于正常组织细胞,然而实验结果却显示癌细胞似乎释放了比正常组织更多的整合事件进入外周血,这一现象引起了发明人的好奇,提示外周血中的hbv整合事件也许具有作为肝癌诊断或预警标志物的潜力,有可能应用于肝脏免疫反应的评估、肝细胞增殖或克隆扩增以及肿瘤进展的监测过程中。
实施例6、慢性乙肝患者cfdna分析
为了验证外周血cfdna中的整合事件与肝癌的发生以及肝癌状态之间是否存在联系,从而为肝癌的诊断和预警提供有用的参考信息,需要确定在外周血、肿瘤组织和正常组织中都存在的18个整合事件,究竟是来源于肿瘤组织,还是来源于正常组织,还是同时来源于二者。为此,提取了10例未患肝癌的慢性乙肝患者的外周血cfdna用于验证,按照与前述实施例中相同的方法进行了捕获和测序,以此来确定正常肝组织(即非肿瘤肝组织)中的整合事件是否会释放到外周血中。
这10例患者(参见以下表格)均经历了长时间hbv感染状态(大于25年,均为出生时即感染),结果显示,无论afp水平高低、hbv病毒复制是否活跃,外周血cfdna中均只能检测到低水平的整合事件。将10例慢肝患者与前一实施例中的7例肝癌患者的cfdna中整合事件的水平进行比较,结果显示二者存在显著性差异(结果如图1所示)。
这一结果证明,在未患癌的慢肝患者中,整合事件是很难释放到外周血中的,前一实施例中外周血cfdna中捕获到的整合事件大多是来源于肝肿瘤细胞的,是因为含有整合事件的肝细胞发生了过量增殖,导致其中的cfdna大量进入血液而来不及清除,从而在血液中检测到高丰度的整合事件。
实施例7、整合事件丰度分析
为了确定能否利用外周血cfdna代替肝组织样品来反映受试者的乙肝病毒整合状况,对外周血中检出的整合位点(图2,白)与肝癌组织中的整合位点(图2,黑)进行了比较,发现二者显示出较为一致的丰度,即在肝癌组织中丰度比较高的整合事件,在血浆中的丰度也相对较高,但整合丰度在肝癌组织和正常组织间存在显著差异(p<0.05),进一步支持cfdna为肿瘤组织来源。由于过量增殖的细胞中的整合事件能够大量进入外周血,而普通细胞中的整合只有少量进入血液,并且整合事件增殖程度越高,cfdna中的丰度也越高,因而外周血cfdna中的乙肝病毒整合情况能够反映受试者的乙肝病毒整合状况。
为了在外周血cfdna的整合事件中寻找能够最好地反映肝细胞过量增殖情况的整合事件,比较了不同的入选标准,选取基因组序列匹配度、乙肝病毒序列匹配度和断点出现率作为指标,每个指标选取数档标准,即整合事件中宿主基因组序列与参比序列之间的核苷酸差异不超过1/2/3/4个,整合事件中乙肝病毒序列与参比序列之间的核苷酸差异不超过3/4/5/6个,同一整合事件每一端的断点都在至少1/2/3个非冗余的跨断点读长(junction)中存在。按照上述标准筛选cfdna中的整合事件后,得到了最佳的筛选条件:整合事件中基因组序列的匹配度不超过2个核苷酸差异,乙肝病毒序列的匹配度不超过5个核苷酸差异,同一整合事件的断点在至少2个非冗余的跨断点读长中存在。如图3所示,在此条件下,可以得到与肿瘤组织断点丰度最佳的相关性(r2=0.62)。
在外周血中的整合事件丰度约为组织中的10%,其实受到肿瘤异质性的影响,切取肿瘤组织并分析里面的整合,不一定与肿瘤中实际的丰度吻合,在此条件的影响下,外周血中的整合覆盖度依旧表现出较好的相关性,考虑到组织取样本身对于肿瘤整体组成反映时带来的抽样偏差,外周血的检测甚至可能比组织取样更好地反映了患者体内肿瘤该克隆的比例。
实施例8、整合事件插入/断裂模式分析
将10例慢肝患者与7例肝癌患者的外周血cfdna中的整合事件进行比较,结果显示二者展现出一致的片段插入/断裂模式(参见图4),但慢肝患者的断点丰度尚未达到可以判断含有整合的肝细胞异常增殖的所需阈值。慢肝患者肝脏内也在时刻发生整合,并且一部分带有整合的细胞死亡后,其dna释放入血后也可能被检测到,在挑选慢肝患者时,着重挑选了肝癌发生风险高、但现有超声诊断技术尚未诊断出肝癌的患者,利用捕获方案发现的整合片段候选区域,展现出与肝癌患者外周血中一致的插入/断裂模式,提示整合的确会时刻发生,但是这种整合片段在外周血中的丰度极其有限,仅有1个整合的嵌合读长,证实释放的整合片段的丰度需要达到一定阈值,即带有整合的肝脏细胞需要增殖到一定程度,才会存在发生肿瘤的可能。通过对受试者外周血cfdna中的整合事件进行监测,发现丰度达到阈值后,可进行肝癌诊断或定期筛查,诊断和筛查可以采用常用的手段,例如影像学检测。
参考文献
1.k.yaginumaetal.jvirol61,1808-1813(1987).
2.t.nagayaetal.genes&development1,773-782(1987).
3.y.shaul,etal.journalofvirology59,731-734(1986).
4.o.hino,etal.pnas83,8338-8342(1986).
5.m.fowleretal.journalofhepatology2,218-229(1986).
6.a.dejean,etal.nature322,70-72(1986).
7.k.yaginuma,etal.pnas82,4458-4462(1985).
8.c.rogleretal.science230,319-323(1985).
9.r.koshyetal.cell34,215-223(1983).
10.c.brechot,etal.nature286,533-535(1980).
11.p.r.chakraborty,etal.nature286,531-533(1980).
12.r.koshyetal.cell34,215-223(1983).
13.d.a.shafritz,etal.nengljmed305,1067-1073(1981).
14.m.d.andrake,etal.annurevvirol2,241-264(2015).
15.t.nagayaetal.genesdev1,773-782(1987).
16.w.yang,etal.jvirol73,9710-9717(1999).
17.m.nassal.gut64,1972-1984(2015).
18.w.li,annurevcelldevbiol31,125-147(2015).
19.w.k.sung,etal.natgenet44,765-769(2012).
20.v.schluter,etal.oncogene9,3335-3344(1994).
21.m.geng,etal.worldjgastroenterol21,10732-10738(2015).
22.t.pollicino,etal.jhepatol61,408-417(2014).
23.h.dongetal.plosone10,e0123175(2015).
24.y.t.chiuetal.jhepatol,(2016).
25.w.s.masonetal.gastroenterology151,986-998e984(2016).
26.c.i.wooddelletal.scitranslmed9(2017).
27.h.yanetal.hepatology61,1821-1831(2015).
28.r.miaoetal.jhepatol61,840-849(2014).
29.m.minami,etal.genomics29,403-408(1995).
30.y.murakamietal.gut54,1162-1168(2005).
31.t.kanagawa,etal.jbioscibioeng96,317-323(2003).
32.m.s.judo,etal.nucleicacidsres26,1819-1825(1998).
33.x.lietal.hepatol60,975-984(2014).
34.g.amaddeoetal.gut64,820-829(2015).
35.t.tu,etal.viruses9,(2017).
36.m.heikenwalder,etal.cellhostµbe15,249-250(2014).
37.z.jiangetal.genomeresearch22,593-601(2012).
38.p.pineauetal.journalofvirology70,7280-7284(1996).
39.s.a.beckeretal.cancerresearch56,5092-5097(1996).
40.m.meyer,etal.hepatology15,665-671(1992).
41.t.tokino,etal.journalofvirology65,6761-6764(1991).
42.h.wang,etal.cytogeneticandgenomeresearch48,72-78(1988).
43.l.-h.zhaoetal.naturecommunications7,12992(2016).
44.c.neuveut,etal.jhepatol52,594-604(2010).
45.m.maetal.anntranslmed3,235(2015).
46.f.lietal.nanfangyikedaxuexuebao34,147-152(2014).
47.s.villaretal.environhealthperspect119,1635-1640(2011).
48.y.zhaoetal.clinepigenetics6,30(2014).
以上所述仅是本发明的优选实施方式,并不用于限定本发明的保护范围,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!