一种阿齐沙坦微粉原料药的制备工艺的制作方法
本发明属于制药工程和药物结晶技术领域,具体涉及一种阿齐沙坦微粉原料药的制备工艺。
背景技术
阿齐沙坦(azilsartan),化学名:2-乙氧基-1-[[2'-(4,5-二氢-5-氧代-1,2,4-恶二唑-3-基)联苯-4-基]甲基]苯并咪唑-7-羧酸,结构如式ⅰ所示。阿齐沙坦是由日本武田(takeda)制药株式会社开发的一款血管紧张素ⅱ受体拮抗剂,2012年在日本获批上市,临床主要用于治疗高血压症。阿齐沙坦的临床疗效和副作用相对于坎地沙坦酯显示出了更强的优势,被视作坎地沙坦酯的下一代产品,具有非常好的市场前景。
阿齐沙坦原料药几乎不溶于水,在生物药剂学(bcs)分类系统中,阿齐沙坦属于第ⅳ类,即低溶解性,低渗透性药物。在药物制剂中,第ⅳ类药物由于溶解度低,溶出速率慢等特点,使得药物溶出度成为限制其人体内生物利用度高低的关键因素。根据noyes-whimey方程,减小药物颗粒度,增大药物比表面积,能有效加快药物的溶出速度。因此,微粉化技术成为解决难溶性药物溶出度的重要途径。在阿齐沙坦制剂的处方工艺中,往往需要使用微粉化的原料药,以减小原料药的粒径,增大原料药比较面积,从而有效地控制制剂中阿齐沙坦的溶出速度。
目前,制药工业中获得微粉化原料药的主要途径有两种,其一是通过微粉机的机械剪切原理实现微粉化粉碎,其二是通过药物结晶工艺达到有效控制晶体粒径的方式实现微粉化。由于市场上可选的微粉机种类较多,微粉机的机械粉碎手段往往是实现原料药微粉化粉碎的最常用手段。而通过药物结晶方式控制药物颗粒度的方式实现微粉化,往往因为技术难度较大而使得很多企业望而却步。
本发明人在对阿齐沙坦原料药微粉化技术进行研究的过程中发现:通过微粉机进行机械粉碎的原料药与微粉前相比,杂质明显增多、增大;且微粉化后的原料药稳定性差,6个月长期稳定性试验和3个月加速稳定性试验均表明,微粉化后的原料药杂质增长趋势明显,并超出ich对原料药中已知杂质控制限度的要求,即已知杂质不得大于0.15%。研究发现,通过机械手段微粉化的原料药中,杂质a和杂质b明显增大,并出现新杂质c,且杂质a、b、c均为阿齐沙坦的降解杂质。这些杂质随着微粉时间的延长也出现明显增大的趋势。在对微粉化原料药进行稳定性研究过程中发现,杂质a和杂质b在6个月长期存放条件和3个月加速存放条件下,均超出0.15%的控制限度,并接近0.30%的水平。这些降解杂质的形成过程和结构如下所示:杂质a由温度过高引起降解;因为阿齐沙坦工艺过程中使用了乙醇,所以高温条件下,残留在原料药中的乙醇参与了反应,形成了杂质b和杂质c。
微粉机对阿齐沙坦原料药进行机械粉碎的过程中杂质明显增多、增大的原因可能是:由于微粉机在对阿齐沙坦原料药进行机械剪切、粉碎的过程中,机械剪切会产生热量,使得新剪切面的温度过高引起原料药的降解。而机械粉碎后的原料药稳定性变差的主要原因可能是:机械粉粹过程中形成了很多新的剪切面,新形成的剪切面表面能较大,使得新剪切面的药物分子能量较高,更容易发生降解。
为了避免类似的情况发生,隋立朋等人在专利cn103831159中公开了一种超低温条件下粉碎阿齐沙坦原料药的方法,该工艺过程仍采用机械粉碎的手段,只是在微粉化过程中使用了液氮降温等措施。虽然该方法解决了原料药过程杂质增大的问题,但机械粉碎后原料药的稳定性仍未得到解决。且该粉碎工艺操作繁琐,需要分多批次加料,粉碎过程中需要不断加入液氮。同时该微粉碎工艺也存在如下缺陷:一方面,液氮超低温度下长期工作大大缩短了微粉机的使用寿命;另一方面,大量液氮的加入后气化,可能使微粉碎过程中气体迅速膨胀,具有安全风险;再者,大量液氮的使用,增大了粉碎空间缺氧窒息的风险,操作人员人身安全风险较高;此外,液氮的使用增加了微粉碎工艺的成本。所以,隋立朋等人在专利cn103831159中公开的工艺,并不适合于产业化。
袁秀菊等人在专利cn103664921中,公开了一种阿齐沙坦晶型a及其制备方法,其所述的结晶颗粒度较小,但结果显示所得阿齐沙坦原料药粒径d(90)仍然在70μm左右,并不符合阿齐沙坦制剂处方工艺中对原料药粒径的要求(d(90)不大于20μm)。
综上所述,机械微粉化手段并不适合于阿齐沙坦原料药的微粉化,而常规的结晶方法难以实现阿齐沙坦原料药的微粉化。鉴于此,本发明提供一种通过溶剂分散和结晶组合的方法控制原料药粒径的大小的工艺,实现了对阿齐沙坦原料药的微粉化。该制备工艺收率高、操作简便、微粉化后的产品稳定性良好,微粉化后的原料药粒径能到达制剂处方工艺中的粒径要求,且适合于工业化生产。
技术实现要素:
本发明提供一种阿齐沙坦微粉原料药的制备工艺,更具体而言,本发明提供了一种通过溶剂分散和结晶组合的方式实现阿齐沙坦原料药微粉化的工艺。该工艺收率高、操作简便、微粉化后的产品稳定性良好,,微粉化后的原料药粒径能到达制剂处方工艺中的粒径要求,且适合于工业化生产。
本发明所述的阿齐沙坦微粉原料药的制备工艺,包括以下内容:
一种阿齐沙坦微粉原料药的制备工艺,其特征包括如下步骤:
(1)阿齐沙坦原料药经有机溶剂a分散;
(2)步骤(1)中分散所得样品,经有机溶剂b重结晶。
进一步地,本发明所述的阿齐沙坦微粉原料药的制备工艺,其特征在于,步骤(1)中所述阿齐沙坦原料药的分散方法包括以下过程:阿齐沙坦原料药在x体积的有机溶剂a中形成悬浊液,加热至温度t1搅拌时间t1后,将悬浊液冷却至温度t2,悬浊液再经固液分离、洗涤、干燥工序。
更进一步地,本发明中所述的阿齐沙坦微粉原料药的制备工艺,其特征在于,步骤(2)中所述的结晶方法包括如下过程:步骤(1)分散所得样品中加入y体积的有机溶剂b,加热至温度t3条件下溶解;然后除去z体积的有机溶剂b;经速度v冷却至温度t4搅拌时间t2后,进一步冷却至温度t5搅拌时间t3,最后经固液分离、洗涤、干燥工序。
本发明中所述阿齐沙坦微粉原料药的制备工艺,其特征在于,步骤(1)中所述的有机溶剂a为四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、乙酸乙酯、甲苯、乙腈中的任一种或任两种溶剂的组合,优选四氢呋喃。
本发明中所述阿齐沙坦微粉原料药的制备工艺,其特征在于,步骤(2)中所述的有机溶剂b为丙酮、丁酮、异丙醇、乙醇、甲醇中的任一种或任两种溶剂的组合,优选丙酮。
本发明中所述的阿齐沙坦原料药分散方法,其特征在于,所述的x体积为3~6体积,优选4体积;所述的温度t1为55℃~75℃,优选60~70℃;所述的温度t2为0℃~40℃,优选20~30℃;所述的时间t1为1~5小时,优选1~2小时。
本发明中所述的结晶方法,其特征在于,所述的y体积为45~65体积,优选55体积;所述的温度t3为50℃~70℃,优选55℃~65℃;所述的z体积为25~30体积,优选27体积;所述的冷却速度v为5℃/小时~15℃/小时,优选10℃/小时;所述的温度t4为10℃~30℃,优选20~25℃;所述的时间t2为1~4小时,优选1~2小时;所述的温度t5优选0~5℃;所述的时间t3为1~4小时,优选1~2小时。
本发明中所述的阿齐沙坦微粉原料药的制备工艺,,其特征在于,所得阿齐沙坦微粉的粒径分布为d(90)<20μm。
本发明中所述的阿齐沙坦微粉原料药的制备工艺,其特征在于,所得阿齐沙坦微粉中单个杂质小于0.10%。
本发明中所述的阿齐沙坦微粉原料药的制备工艺,其特征在于,所得阿齐沙坦微粉在温度25±2℃,湿度60±5%rh条件下存放6个月,杂质不出现明显增大。
需要强调和说明的是,本发明中所述的阿齐沙坦微粉原料药的制备工艺是由溶剂分散和重结晶两个过程组合而成。任何通过调整分散溶剂或结晶溶剂的做法,都被视为包括在本发明之内。任何对温度和时间参数进行调整的做法,均被视为包括在本发明之内。
进一步需要强调的是,本发明中所述的步骤(2)中“除去z体积的有机溶剂b”的方法优选但不限于常压蒸馏,本发明中所述的固液分离方法包括但不限于离心、过滤、减压抽滤或压滤等常用方式。
本发明中所述的阿齐沙坦微粉原料药的制备工艺,所得阿齐沙坦微粉的粒径分布为d(90)<20μm是本发明的重要结果性特征,d(90)的大小可以通过结晶过程的冷却速度调节。结晶过程中的冷却速度是控制颗粒度的关键因素。所以,d(90)分布在20μm范围内均应该视为本发明的重要结果性特征。
更进一步需要强调的是本发明中所述的“温度25±2℃,湿度60±5%rh条件下存放6个月”是参照《中国药典》2010版对原料药稳定性考察的一种方式,其结果是用于证明本发明所公开的工艺制备的阿齐沙坦微粉原料药稳定性是否良好,不以任何形式限制本发明的特征,其他形式的稳定性考察试验均能得到相同的结论。
本发明中所涉及到阿齐沙坦的杂质和纯度hplc纯度检测方法如下所述:层析柱用十八烷基硅烷键合硅胶为填充剂(capcellpakc184.6×150mm3μm或等效柱);以10mmol/l磷酸二氢钠溶液(用磷酸调节ph值至3.0)溶液为流动相a;以乙腈为流动相b;梯度洗脱为:0-5min,65%a﹕35%b;25-35min,30%a﹕70%b;35-45min,65%a﹕35%b;流速为每分钟1.0ml;检测波长为250nm,进样量为20μl。
术语:
本发明中所述的“原料药”是指在用于药品制造中的任何一种物质或物质的混合物,而且在用于制药时,成为药品的一种活性成分。此种物质在疾病的诊断,治疗,症状缓解,处理或疾病的预防中有药理活性或其他直接作用,或者能影响机体的功能或结构。
本发明中所述的“ich”是指人用药物注册技术要求国际协调会,是internationalconferenceonharmonizationoftechnicalrequirementsforregistrationofpharmaceuticalsforhμmanuse的英文缩写。
本发明中所述的“d(90)”常用来表示粉体粗端的粒度指标,是指一个样品的累计粒度分布数达到90%时所对应的粒径。它的物理意义是粒径小于它的颗粒占90%。
附图说明
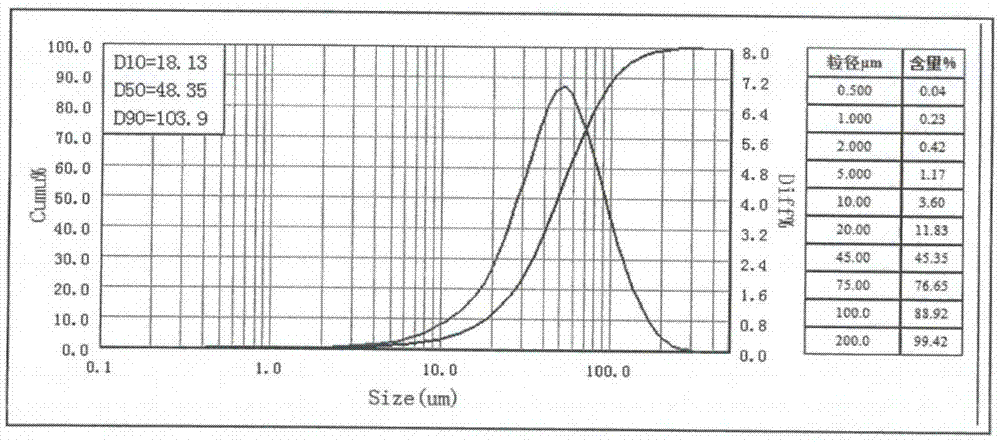
图1微粉前阿齐沙坦原料药粒径分布图
图2分散结晶后阿齐沙坦原料药粒径分布图
图3分散结晶后阿齐沙坦xrd图
图4微粉前阿齐沙坦hplc图
图5机械微粉后阿齐沙坦hplc图
图6分散结晶后阿齐沙坦hplc图
图7机械微粉后6个月稳定性hplc图
图8分散结晶后阿齐沙坦6个月稳定性hplc图
具体实施方式
下面具体实施例可以使本专业技术人员全面理解本发明,但不以任何方式限制本发明。
在下列实施例中,除非另有指明,所有温度为摄氏温度;除非另有指明,所述的室温均为20-30℃;除非另有指明,各种起始原料和试剂均来自市售,均不经进一步纯化直接使用;除非另有指明,各种溶剂均为工业级溶剂,不经进一步处理直接使用;除非另有指明,市售厂家包括但不限于杭州化学试剂、国药试剂等。
下实施例中,均使用相同批次的阿齐沙坦原料药,其纯度和颗粒度数据如下表所示:
a、机械粉碎工艺
实施例1:
取100g阿齐沙坦原料药置于rf-08型高速粉碎机中,间歇性粉碎2分钟,所得阿齐沙坦微粉样品hplc和颗粒度如下表所示:
实施例2:
取100g阿齐沙坦原料药置于rf-08型高速粉碎机中,间歇性粉碎5分钟,所得阿齐沙坦微粉样品hplc和颗粒度如下表所示:
b、本发明中所述的工艺
实施例3:
将25.0g阿齐沙坦原料药、100ml四氢呋喃投入反应瓶中,加热至60~70℃搅拌1h,然后将悬浊液放置降温至20~30℃,过滤,滤饼用50ml四氢呋喃洗涤,得到的滤饼在30~40℃减压干燥,得到分散后的阿齐沙坦26.1g。将分散后的阿齐沙坦、1430ml丙酮投入2l的反应瓶中,加热至55~65℃溶解,然后蒸馏出620ml丙酮,蒸馏结束后,以10℃/小时速率降温至25~30℃,并在25~30℃保温搅拌1小时,随后降温至0~5℃,并在0~5℃保温搅拌2小时,过滤,滤饼用50ml冷的丙酮洗涤,30~40℃真空干燥得到阿齐沙坦23.7g,收率94.8%。hplc纯度和粒径大小如下表所示:
实施例4:
将25.0g阿齐沙坦原料药、100ml四氢呋喃投入反应瓶中,加热至60~70℃搅拌1h,然后将悬浊液放置降温至20~30℃,过滤,滤饼用50ml四氢呋喃洗涤,得到的滤饼在30~40℃减压干燥,得到分散后的阿齐沙坦26.3g。将分散后的阿齐沙坦、1435ml丙酮投入2l的反应瓶中,加热至55~65℃溶解,然后蒸馏出625ml丙酮,蒸馏结束后,以15℃/小时速率降温至20~25℃,并在20~25℃保温搅拌2小时,随后降温至0~5℃,并在0~5℃保温搅拌2小时,过滤,滤饼用50ml冷的丙酮洗涤,30~40℃真空干燥得到阿齐沙坦23.9g,收率95.6%。hplc纯度和粒径大小如下表所示:
实施例5:
将25.0g阿齐沙坦原料药、100ml四氢呋喃投入反应瓶中,加热至回流搅拌1h,然后将悬浊液放置降温至20~30℃,过滤,滤饼用50ml四氢呋喃洗涤,得到的滤饼在30~40℃减压干燥,得到分散后的阿齐沙坦26.0g。将分散后的阿齐沙坦、1430ml丙酮投入2l的反应瓶中,加热至55~65℃溶解,然后蒸馏出620ml丙酮,蒸馏结束后,以5℃/小时速率降温至20~25℃,并在20~25℃保温搅拌1小时,随后降温至0~5℃,并在0~5℃保温搅拌1小时,过滤,滤饼用50ml冷的丙酮洗涤,30~40℃真空干燥得到阿齐沙坦23.0g,收率92.0%。hplc纯度和粒径大小如下表所示:
实施例6:
将25.0g阿齐沙坦原料药、150ml甲基叔丁基醚投入反应瓶中,加热至55~60℃搅拌1h,然后将悬浊液放置降温至0~10℃,过滤,滤饼用25ml冷的甲基叔丁基醚洗涤,得到的滤饼在30~40℃减压干燥,得到分散后的阿齐沙坦25.0g。将分散后的阿齐沙坦、1240ml甲醇投入2l的反应瓶中,加热至60~70℃溶解,然后蒸馏出740ml甲醇,蒸馏结束后放置冷却,以10℃/小时速率降温至20~25℃,并在20~25℃保温搅拌2小时,随后降温至0~5℃,并在0~5℃保温搅拌3小时,过滤,滤饼用50ml冷的甲醇洗涤,30~40℃真空干燥得到阿齐沙坦22.3g,收率89.2%.hplc纯度和粒径如下表所示:
实施例7:
将25.0g阿齐沙坦原料药、100ml2-甲基四氢呋喃投入反应瓶中,加热至70~75℃搅拌1h,然后将悬浊液放置降温至20~30℃,过滤,滤饼用50ml2-甲基四氢呋喃洗涤,得到的滤饼在30~40℃真空干燥,得到分散后的阿齐沙坦27.2g。将分散后的阿齐沙坦、1430ml丁酮投入2l的反应瓶中,加热至60~70℃溶解,然后蒸馏出720ml丁酮,蒸馏结束后,以15℃/小时速率降温至20~25℃,并在20~25℃保温搅拌2小时,随后降温至0~5℃,并在0~5℃保温搅拌2小时,过滤,滤饼用50ml冷的丁酮洗涤,30~40℃真空干燥得到阿齐沙坦21.6g,收率86.4%。hplc纯度和粒径如下表所示:
实施例8:
将25.0g阿齐沙坦原料药、100ml乙酸乙酯投入反应瓶中,加热至60~70℃搅拌1h,然后将悬浊液放置降温至10~20℃,过滤,滤饼用50ml乙酸乙酯洗涤,得到的滤饼在30~40℃减压干燥,得到分散后的阿齐沙坦25.8g。将分散后的阿齐沙坦、1430ml丙酮投入2l的反应瓶中,加热至55~65℃溶解,然后蒸馏出620ml丙酮,蒸馏结束后,以5℃/小时速率降温至25~30℃,并在25~30℃保温搅拌3小时,随后降温至0~5℃,并在0~5℃保温搅拌2小时,过滤,滤饼用50ml冷的丙酮洗涤,30~40℃真空干燥得到阿齐沙坦22.6g,收率90.4%。hplc纯度和粒径如下表所示:
实施例9:
将25.0g阿齐沙坦原料药、125ml甲苯投入反应瓶中,加热至60~70℃搅拌1h,然后将悬浊液放置降温至25~35℃,过滤,滤饼用50ml甲苯洗涤,得到的滤饼在40~50℃真空干燥,得到分散后的阿齐沙坦28.3g。将分散后的阿齐沙坦、1640ml异丙醇投入2l的反应瓶中,加热至60~70℃溶解,然后蒸馏出820ml异丙醇,蒸馏结束后,以15℃/小时速率降温至20~25℃,并在20~25℃保温搅拌1小时,随后降温至0~5℃,并在0~5℃保温搅拌1小时,过滤,滤饼用50ml冷的异丙醇洗涤,40~50℃真空干燥得到阿齐沙坦21.8g,收率87.2%。hplc纯度和粒径如下表所示:
实施例10:
将25g阿齐沙坦原料药、100ml乙腈投入反应瓶中,加热至60~70℃搅拌1h,然后将悬浊液放置降温至20~30℃,过滤,滤饼用50ml乙腈洗涤,得到的滤饼在30~40℃减压干燥,得到分散后的阿齐沙坦24.6g。将分散后的阿齐沙坦、1430ml乙醇投入2l的反应瓶中,加热至60~70℃溶解,然后蒸馏出680ml乙醇,蒸馏结束后,以10℃/小时速率降温至20~25℃,并在20~25℃保温搅拌3小时,随后降温至0~5℃,并在0~5℃保温搅拌4小时,过滤,滤饼用50ml冷的乙醇洗涤,30~40℃真空干燥得到阿齐沙坦22.5g,收率90.0%。hplc纯度和粒径如下表所示:
c、稳定性试验
实施例11:
取实施例1中所得阿齐沙坦微粉原料药1.0g,双层pe袋,一层铝箔袋包装好后放置于温度25±2℃,湿度60±5%rh条件的稳定性研究箱中,6个月后取样进行hplc检测,结果如下表所示:
结论:以上结果显示,实施例1中所得微分原料药在温度25±2℃,湿度60±5%rh条件下存放6个月,稳定性较差,纯度明显降低,杂质明显增大。
实施例12:
取实施例3中所得阿齐沙坦微粉原料药1.0g,双层pe袋,一层铝箔袋包装好后放置于温度25±2℃,湿度60±5%rh条件的稳定性研究箱中,6个月后取样进行hplc检测,结果如下表所示:
结论:以上结果显示,实施例3中所得微粉原料药在温度25±2℃,湿度60±5%rh条件下存放6个月,稳定性较好,纯度未见明显降低,杂质未见明显增大。
本发明的方法通过较佳实施例进行了描述,相关领域人员明显能在本发明内容和范围内对本发明中所述的方法和应用在有必要的地方稍加适当常识性的调整、改动和组合,来实现和应用本发明技术。本领域人员也可以借鉴本发明内容,通过适当改进工艺参数实现。特别需要指出的是,所有类似的改进和调整对本领域技术人员来说是显而易见的,都应被视为包括在本发明之内。
- 还没有人留言评论。精彩留言会获得点赞!