与I型神经纤维瘤合并脊柱畸形病相关GPR56及其应用的制作方法
本发明涉及生物技术和医学检测
技术领域:
,具体涉及与i型神经纤维瘤合并脊柱畸形病相关gpr56及其应用。
背景技术:
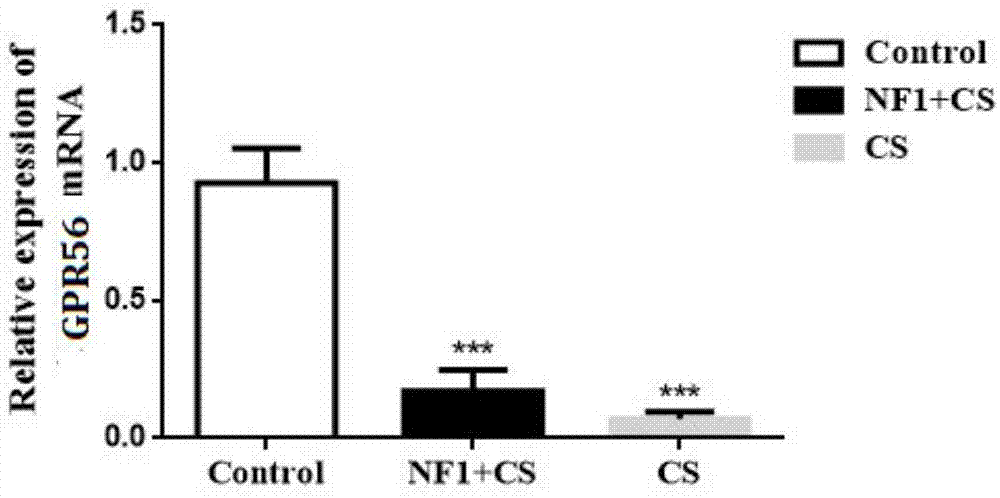
:i型神经纤维瘤病(neurofibromatosistype1,nf1)是一种较为常见的常染色体显性遗传病;不分性别或种族,则全球的发病率1/3000。1882年frederichvonrecklinghausen首次描述了nf1,1987年nih发表了正式的诊断标准。遗传学研究发现,i型神经纤维瘤病是由nf1肿瘤抑制基因的突变所致。nf1基因位于第17染色体长臂17q11.2,编码分子量为220kda的胞浆蛋白——神经纤维素蛋白(neurofibromin),该蛋白的部分作用为负向调控ras原癌基因,而ras是调控细胞生长的重要信号分子。基因突变的患者,体内每个细胞都含有一个突变无功能的nf1拷贝和一个功能正常的nf1拷贝。nf1型患者易发生中枢和周围神经系统的良、恶性肿瘤,以及其它部位的恶性病变。与nf1相关的肿瘤常见的包括:视觉通路的神经胶质瘤、胶质母细胞瘤、恶性周围神经鞘瘤、胃肠道间质瘤、乳腺癌、白血病、嗜铬细胞瘤、十二指肠类癌瘤和横纹肌肉瘤。尽管此病的许多临床特征从患者出生后就很明显,但形成肿瘤却需要某些细胞获得性nf1变异,完全丧失基因功能。约有50%的nf1患者并无家族史,此病为自发性基因突变。随着基因检测技术的发展,基因型-表现型的关系已经大量被研究。譬如,nf1基因微缺失的患者表现型更为严重,易在早年就出现神经纤维瘤、平均iq较低、面部特征异常、发生恶性周围神经鞘瘤的风险增加。先天性脊柱侧凸(congenitalscoliosis,cs)是一种常见的脊椎疾病,新生儿患病几率为0.5-1‰。cs的临床表现为脊柱侧弯超过10度,这是由于胚胎的发育过程中脊柱畸形(半椎体畸形、分节障碍、蝴蝶椎、并肋等)引起的脊柱纵向生长不平衡导致的。cs可以影响生理和心理的健康,已经成为青少年残疾的一个主要的因素。以往认为,大多数先天性脊柱侧凸是非遗传性的,是胚胎在发育过程中由环境因素引起的。近年来,已有研究表明遗传因素参与了cs的发病机制。以前在动物模型中的基因操作实验表明遗传缺陷导致了脊椎异常。有趣地是,已表明人类基因(例如,dll3,hes7,mesp2和t)的一些突变参与了cs的过程;然而,这些突变可从表型正常的家族成员那遗传而来。家族内部相同突变导致的不同表型的差异说明了人类cs遗传变异的复杂性。基因与环境的相互作用已被提出用于解释上述现象。自1983年至今北京协和医院骨科已收集神经纤维瘤病脊柱畸形样本178例,通过多种技术手段,对这些患者的临床信息、影像学资料(完整的x线、mri和ct)及随访情况进行分析,发现其中非营养不良性脊柱侧凸患者42例(23.6%),营养不良性脊柱侧凸患者136例(76.4%)。营养不良性脊柱侧凸患者不仅表现脊柱侧凸,而且具有严重的骨骼发育异常,其中有9例出现严重的肋椎关节脱位,肋骨突入椎管。同时营养不良性脊柱侧凸患者矫形手术后易出现融合失败、假关节形成等并发症,发生率高达25(18.4%),矫形手术失败率远远高于先天性脊柱侧凸患者。由此可见,i型神经纤维瘤合并脊柱畸形是多系统的综合疾病,尤其以营养不良性i型神经纤维瘤合并脊柱侧凸为代表,此类患者术后易发生骨愈合缺陷,造成矫形手术失败,对患者的身体健康和生活质量造成极大影响。因此,深入探讨营养不良性i型神经纤维瘤合并脊柱侧凸相关的发病机制,发现造成此类患者骨骼愈合缺陷、矫形手术失败的根本性原因,并制定相应的治疗方案介入矫形手术和患者的术后康复,不仅可以推动i型神经纤维瘤合并脊柱畸形这类疾病的基础理论认识,同时相应治疗方案的改进对此类患者临床矫形手术成功率的提高具有十分重要的意义。鉴于目前还没有与i型神经纤维瘤合并脊柱畸形病产品早期诊断诊断产品,如能发现相应的标记物和研制相应的诊断试剂盒,将为探索i型神经纤维瘤合并脊柱畸形病早期防治的药物靶点提供新的方向,并为其药物筛选、药效评价及靶向治疗开辟新的途径。技术实现要素:本发明的目的在于寻找一种与i型神经纤维瘤合并脊柱畸形病相关的生物标志物—gpr56以及提供所述标志物在制备i型神经纤维瘤合并脊柱畸形病检测或治疗产品中的应用;本发明的另一目的在于阐明gpr56与i型神经纤维瘤合并脊柱畸形病发生的分子机制,寻找预防或治疗i型神经纤维瘤合并脊柱畸形病的新途径。本发明的目的是通过下列技术方案实现的:发明人通过10例(5&5)nf1合并脊柱畸形患者转录组测序,筛选到新的潜在关键基因—gpr56,并通过westernblot验证gpr56蛋白在患者外周血中表达显著下调。进一步,发明人分别利用稳定敲低grp56的神经纤维瘤细胞、成骨细胞和破骨细胞作为研究模型,观察敲低grp56对细胞增殖、周期、以及对成骨细胞、破骨细胞分化相关标志物的影响。首先,本发明提供了gpr56作为标志物在如下任一项所述中的应用:1)在制备检测i型神经纤维瘤合并脊柱畸形病的产品中的应用;2)在制备治疗i型神经纤维瘤合并脊柱畸形病的药物中的应用;3)在筛选预防或治疗i型神经纤维瘤合并脊柱畸形病的候选化合物中的应用;4)在抑制骨吸收产品中的应用。优选的,所述检测,包括:a)鉴定受试者的样本中的标志物,其中所述标志物为gpr56;以及b)将所述标志物与参照进行比较,其中与参照相比的所述标志物的表达差异用于检测i型神经纤维瘤合并脊柱畸形病。优选的,所述样本为血液或细胞。优选的,所述产品包括芯片、制剂或试剂盒。优选的,所述gpr56表达水平在i型神经纤维瘤合并脊柱畸形患者的样本中表达水平下调。优选的,所述gpr56在检测i型神经纤维瘤合并脊柱畸形病产品中作为标志物的核苷酸序列如seqidno:1所示。优选的,所述筛选预防或治疗i型神经纤维瘤合并脊柱畸形病的候选化合物可以利用gpr56表达情况来进行筛选。在本发明的一种实施方式中,包括步骤:测试组中,在培养体系中添加测试化合物,并观察所述测试组的细胞中gpr56的表达量和/或活性;在对照组中,在相同的培养体系中不添加测试化合物,并观察对照组的所述细胞中gpr56的表达量和/或活性;本发明可以利用gpr56筛选预防或治疗i型神经纤维瘤合并脊柱畸形的化合物,其中,如果测试组中细胞的gpr56的表达量和/或活性高于对照组,就表明该测试化合物是对gpr56的表达和/或活性有促进作用的治疗疾病的候选化合物。作为本发明的一种实施方式,所述的步骤还包括:对获得的候选化合物进行进一步的细胞实验和/或动物试验,以从候选化合物中进一步选择和确定对于预防、缓解或治疗i型神经纤维瘤合并脊柱畸形有用的物质。作为本发明的实施方式,筛选预防或治疗i型神经纤维瘤合并脊柱畸形的候选化合物的体系不限于细胞体系,还包括亚细胞体系、溶液体系、组织体系、器官体系或动物体系等,所述体系不局限于上述形式,只要所述体系可以检测测试化合物可以升高gpr56的表达和/活性即可。进一步地,本发明提供了一种检测i型神经纤维瘤合并脊柱畸形病的试剂盒,其包括检测样本中gpr56基因或其蛋白表达水平的试剂。优选的,所述试剂选自:特异性识别gpr56基因的探针;或特异性扩增gpr56基因的引物;或特异性结合gpr56编码的蛋白的抗体或配体。优选的,所述特异性扩增gpr56基因的引物序列如seqidno.2~3所示。发明人利用sirna敲低神经纤维瘤细胞中gpr56表达后,明显促进神经纤维瘤细胞增殖,并改变神经纤维瘤细胞周期分布,影响细胞周期进程。发明人利用sirna敲低成骨细胞中gpr56表达后,伴随有成骨分化相关标志物opn(骨桥蛋白)、ocn(细胞骨钙素)、alp(碱性磷酸酶)的表达下降。其中,alp和opn被认为是成骨分化的早期标志物,ocn是成骨细胞分化成熟的标志物。alp会促进体内的钙化过程,它的水平可以反映成骨细胞分化的活跃状况,alp活性越高则证明成骨细胞分化程度越高;opn在骨组织成熟过程中能抑制结晶体的生长,是成骨细胞基质形成和进入矿化成熟时期的标志;ocn通常在骨细胞分化的终末时期产生,它标志着成骨分化成熟。发明人利用sirna敲低破骨细胞中gpr56表达后,与破骨相关标志物nfatc1(活化t细胞核因子1)、c-fos(原癌基因)表达下调。破骨细胞直接参与骨吸收,是骨组织吸收的主要功能细胞。nfatc1可调控tartrateresistant(抗酒石酸盐)在破骨细胞内的表达,对破骨细胞黏附、融合及骨吸收发挥着重要作用。在破骨细胞分化中,nfatc1的作用至关重要;缺乏nfatc1的胚胎干细胞,即使在核因子nf-kβ受体配体(nuclearfactorkappaβligand,rankl)及巨噬细胞集落刺激因子存在的情况下,也不能形成破骨细胞。而过表达nfatc1的骨髓基质细胞则在rankl缺乏的条件下,依然能向破骨细胞分化。而c-fos同样是破骨细胞分化过程中的重要转录因子。因此,寻找到了预防或治疗i型神经纤维瘤合并脊柱畸形病的新途径—促进gpr56基因或蛋白的过表达。在本发明中,所述rna干扰(rnainterference,rnai)是指在进化过程中高度保守的、由双链rna(double-strandedrna,dsrna)诱发的、同源mrna高效特异性降解的现象。使用rnai技术可以特异性剔除或关闭特定基因的表达,该技术已被广泛用于探索基因功能和传染性疾病及恶性肿瘤的基因治疗领域。以细胞为基础的rnai筛选在功能基因学研究方面具有许多优势,主要表现在大多数细胞类型都能使用rnai方法,并且相对较容易下调或沉默任何目的基因的表达。为了确保基因能够被高效剔除或沉默,根据基因的mrna序列设计了sirna特异性片段。sirna的设计根据已发表的通用设计原则(elbashiret.al2001,schwarzet.al2003,khvorovaet.al2003,reynoldset.al2004,hsiehet.al2004,ui-teiet.al2004),通过在线工具完成设计,该在线工具为:sirnaselectionprogramofwhiteheadinstitute(bingbingyuanet.al2004,http://jura.wi.mit.edu/bioc/sirnaext/)和block-ittmrnaidesignerofinvitrogen(winnerofthe2004frost&sullivanexcellenceinresearchaward,https://rnaidesigner.invitrogen.com/sirna/)。为了进一步提高sirna片断的有效性,综合两个在线设计工具的优点来设计用于筛选的sirna片断。最后,通过同源性比对(ncbiblast)来过滤sirna序列,以提高sirna片断的特异性并减少rnai干扰的脱靶效应。进一步地,本发明还提供了gpr56基因和/或其编码的蛋白的促进剂在制备预防或治疗i型神经纤维瘤合并脊柱畸形病的药物组合物中的应用。优选的,所述促进剂包括促进gpr56基因或其编码的蛋白表达水平、促进gpr56表达功能、和/或促进gpr56表达产物活性的试剂。通常,可将这些促进剂配制于无毒的、惰性的和药学上可接受的水性载体介质中,其中ph通常约为5-8,较佳地ph约为6-8,尽管ph值可随被配制物质的性质以及待治疗的病症而有所变化。配制好的药物组合物可以通过常规途径进行给药,其中包括(但并不限于):瘤内、肌内、腹膜内、静脉内、皮下、皮内、或局部给药。作为本发明的一种优选方式,所述的gpr56的促进剂是一种gpr56的表达载体。所述的表达载体通常还含有启动子、复制起点和/或标记基因等。本领域的技术人员熟知的方法能用于构建本发明所需的表达载体。这些方法包括体外重组dna技术、dna合成技术、体内重组技术等。所述的表达载体优选地包含一个或多个选择性标记基因,以提供用于选择转化的宿主细胞的表型性状,如卡拉霉素、庆大霉素、潮霉素、氨苄青霉素抗性。在本发明中,是本领域已知的各种载体,如市售的载体、包括质粒、粘粒、噬菌体、病毒等。表达载体向宿主细胞中的导入可以使用电穿孔法、磷酸钙法、脂质体法、deae葡聚糖法、显微注射、病毒感染、脂质体转染、与细胞膜透过性肽的结合等周知的方法。在本发明中,“宿主细胞”可以是原核细胞,如细菌细胞;或是低等真核细胞,如酵母细胞;或是高等真核细胞,如哺乳动物细胞。代表性例子有:大肠杆菌,链霉菌属的细菌细胞;真菌细胞如酵母;植物细胞;果蝇s2或sf9的昆虫细胞;cho、cos、或293细胞的动物细胞等。用重组dna转化宿主细胞可用本领域技术人员熟知的常规技术进行。当宿主为原核生物如大肠杆菌时,能吸收dna的感受态细胞可在指数生长期后收获,用cacl2法处理,所用的步骤在本领域众所周知。另一种方法是使用mgcl2。如果需要,转化也可用电穿孔的方法进行。当宿主是真核生物,可选用如下的dna转染方法:磷酸钙共沉淀法,常规机械方法如显微注射、电穿孔、脂质体包装等。优选的,所述药物组合物包括gpr56的促进剂,和/或与所述促进剂配伍的其他药类以及药学上可接受的载体和/或辅料。优选的,所述药学上可接受的载体和/或辅料,包括(但不限于)稀释剂、粘合剂、表面活性剂、致湿剂、吸附载体、润滑剂、填充剂、崩解剂。本发明中的药物组合物还可以包括稳定剂、杀菌剂、缓冲剂、等渗剂、螯合剂、ph控制剂及表面活性剂等添加剂。本发明所述药物组合物可口服给药、非胃肠道给药、通过吸入喷雾给药、局部给药、直肠给药、鼻给药、颊给药、阴道给药或通过植入的贮药装置给药。优选口服给药或注射给药。本发明的药物组合物还可以与其他治疗i型神经纤维瘤合并脊柱畸形病的药物联用,其他治疗性药物可以与主要的活性成分同时给药,甚至在同一组合物中同时给药。还可以以单独的组合物或与主要的活性成分不同的剂量形式单独给予其它治疗性药物。优选的,可采用基因治疗的手段进行。比如,可直接将gpr56的促进剂通过诸如注射等方法给药于受试者;或者,可通过一定的途径将携带gpr56的促进调剂的表达单位(比如表达载体或病毒等)递送到靶点上,具体情况需视所述的促进剂的类型而定,这些均是本领域技术人员所熟知的。本发明有益效果:本发明通过转录组测序技术筛选到nf1合并脊柱畸形相关标记物gpr56,所述gpr56在nf1合并脊柱畸形病患者的样本中表达显著下调,并简单阐明了gpr56在nf1合并脊柱畸形病的作用机理,通过下调gpr56,促进了神经纤维瘤细胞的增殖,抑制成骨分化相关基因的表达,同时也抑制了破骨细胞分化相关标志物的表达,为指导其临床早期干预及靶向治疗提供了重要理论依据,为nf1合并脊柱畸形病提供一种新的治疗途径—gpr56促进剂。进一步本发明将gpr56作为nf1合并脊柱畸形新型生物标志物在制备检测nf1合并脊柱畸形的产品、预防或治疗nf1合并脊柱畸形药物中的应用以及在筛选预防或治疗i型神经纤维瘤合并脊柱畸形病的候选化合物中的应用。附图说明图1本发明gpr56基因cdna在各组中的表达;图2利用westernblot检测各组中gpr56蛋白表达情况;图3利用westernblot检测sirna转染对rsc96细胞中gpr56蛋白表达的影响;图4检测敲低gpr56后对rsc96细胞增殖的影响;图5检测敲低gpr56后对rsc96细胞周期的影响;图6利用westernblot检测sirna转染后对成骨细胞中gpr56蛋白表达的影响;图7利用westernblot检测gpr56敲低后,对成骨细胞分化相关的基因表达的影响;图8利用westernblot检测sirna转染对破骨细胞中gpr56蛋白表达的影响;图9利用westernblot检测gpr56敲低后,对破骨细胞相关的标志物表达的影响。具体实施方式以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。本发明中术语解释如下:fpkm(fragmentsperkilobasespermillionreads)是每百万reads中来自某一基因每千碱基长度的reads数目。在本发明中,“芯片”、“微阵列”、“阵列”可以等同替代,包括(但不限于):dna微阵列(例如,cdna微阵列和寡核苷酸微阵列)、蛋白质微阵列、组织微阵列、转染或细胞微阵列、化学化合物微阵列和抗体微阵列。通常称为基因芯片、dna芯片或生物芯片的dna微阵列是微观dna点的集合,这些点连接到固体表面(例如,玻璃、塑料或硅芯片)上,形成用于对数千种基因同时进行表达谱分析或表达水平监测的阵列。固定的dna片段称为探针,其数千个可用于单个dna微阵列中。微阵列可用于通过比较疾病和正常细胞中的基因表达而识别疾病基因或转录本(例如,ncrna)。微阵列可使用多种技术加以制造,包括但不限于:用细尖针印刷到载玻片上、使用预制的掩模进行光刻、使用动态微镜器件进行光刻、喷墨印刷或微电极阵列上的电化学方法。术语“探针”指能与另一分子的特定序列或亚序列或其它部分结合的分子。除非另有指出,术语“探针”通常指能通过互补碱基配对与另一多核苷酸(往往称为“靶多核苷酸”)结合的多核苷酸探针。根据杂交条件的严格性,探针能和与该探针缺乏完全序列互补性的靶多核苷酸结合。探针可作直接或间接的标记,其范围包括引物。杂交方式,包括,但不限于:溶液相、固相、混合相或原位杂交测定法。本发明中针对gpr56基因的寡核苷酸探针可以是dna、rna、dna-rna嵌合体、pna或其它衍生物。由于不同的探针长度对杂交效率、信号特异性有不同的影响,所述探针的长度通常至少是14个碱基对,最长一般不超过30个碱基对,与目的核苷酸序列互补的长度以15-25个碱基对最佳。所述探针自身互补序列最好少于4个碱基对,以免影响杂交效率。在本发明中,与gpr56特异性结合的抗体或配体包括单克隆抗体、多克隆抗体;本发明不仅包括完整的抗体分子,也包括抗体的任何片段或修饰,例如,嵌合抗体、scfv、fab、f(ab')2、fv等。只要所述片段能够保留与gpr56蛋白的结合能力即可。用于蛋白质水平的抗体的制备时本领域技术人员公知的,并且本发明可以使用任何方法来制备所述抗体。本发明提供了一种试剂盒,所述试剂盒可用于检测gpr56的表达。优选的,所述的制剂或试剂盒中还含有用于标记rna样品的标记物,以及与所述标记物相对应的底物。此外,所述的试剂盒中还可包括用于提取rna、pcr、杂交、显色等所需的各种试剂,包括但不限于:抽提液、扩增液、杂交液、酶、对照液、显色液、洗液等。本发明的试剂盒中还可附有试剂盒的使用说明书,其中记载了如何采用试剂盒进行检测,和如何利用检测结果对nf1合并脊柱畸形进行判断、对治疗方案进行选择。本发明中,术语“样本”以其最广泛的含义使用。意在包括从任何来源获得的标本或培养物,以及生物和环境样本。生物样本可获自动物(包括人)并涵盖液体、固体、组织和气体。生物样本包括血液制品,诸如血浆、血清等。然而,此类样本不应解释为限制适用于本发明的样本类型。在本发明的具体实施例中,实验都是按照至少重复3次来完成的,结果数据都是以平均值±标准差的方式来表示,采用spss18.0统计软件来进行统计分析的,患者样本与正常对照的配对比较采用t检验,配对样本采用t检验,多样本采用均数的方差检验(anova)进行分析,认为当p<0.05时具有统计学意义。以下实施例中,所用试剂和材料均可商购获得,主要试剂和材料为:小鼠单核巨噬细胞白血病细胞raw264.7、小鼠胚胎成骨细胞mc3t3、大鼠雪旺细胞rsc96均购自国家实验细胞资源共享平台,并按照相关培养方法进行培养、传代以及冻存。本发明rnai干扰rsc96细胞中gpr56表达时使用的sirna-1(sig1116105418)、sirna-2(sig180416015851)、sirna-3(sig180416015857)和阴性对照nc(sin05815122147);以及rnai干扰成骨细胞或破骨细胞中gpr56表达使用的sirna-1(sig1116105406)、sirna-2(sig1116105418)、sirna-3(sig1116105429)以及阴性对照nc(sin05815122147)均购自锐博生物。本发明使用gpr56、alp、opn、ocn、nfatc1、c-fos一抗以及二抗购自abcam公司;使用ribofecttmcp转染试剂购自锐博生物;细胞周期检测试剂盒购自南京凯基生物技术股份有限公司。实施例1样品的收集获取自2017年1月至2017年12月于北京协和医院接受神经纤维素瘤合并脊柱畸形手术或检查的5例患者的外周血和5例相对正常的外周血样本。所有患者术前均未接受化疗或放疗,术前已取得患者的知情同意,并通过伦理委员会审核。样本处理:将edta抗凝全血按1:1比例与trizol混合,充分混匀后置于1.8ml细胞冻存管中,在液氮中迅速冷却30s后置于-80℃的冰箱保存。实施例2外周血rna提取1、匀浆处理取新鲜的血液,加入3倍体积红细胞裂解液,混匀后室温放置10min,10000rpm离心1min。弃上清,收集白细胞沉淀。每100-200μl血液收集的白细胞沉淀加入1mltrizol。2、分层样本加入trizol后,室温放置5min,使样本充分裂解。每1mltrizol加入200μl氯仿,剧烈震荡混匀后室温放置3-5min使其自然分相。3、rna沉淀4℃12000rpm离心10-15min。样品会分为3层:黄色有机相,中间层和无色水相,rna主要在水相中,把水相(通常可吸取550-600μl)转移到新ep管中。注:小心吸取水相,千万不要吸取中间界面,否则将导致rna样品中有dna污染。在上清中加入等体积事先预冷的异丙醇,室温放置10-20min。4℃12000rpm离心10min,弃上清,rna沉淀于管底。4、rna漂洗rna沉淀中加入1ml75%乙醇(用rnase-free水配制),温和振荡离心管,悬浮沉淀。没1mltrizol加入1ml75%乙醇。4℃5000-8000rpm离心1-2min,弃上清;可短暂快速离心,用移液器小心吸弃上清,注意不要吸弃沉淀。室温放置1-2min晾干沉淀。5、rna溶解沉淀中加入50-100μlrnase-free水,轻弹管壁,以充分溶解rna,-80℃保存。6、rna完整性及纯度检测完整性:rna可用普通琼脂糖凝胶电泳(电泳条件:1.2%胶;0.5×tbe电泳缓冲液;150v,15min)检测完整性。rna样本中最大rrna亮度应为次大rrna亮度的1.5-2.0倍,否则表示rna样品的降解。出现弥散片状或条带消失表明样品严重降解。纯度:od260/od280比值是衡量rna样品中蛋白质污染程度的指标。高质量的rna样品,od260/od280比值(10mmtris,ph7.5)在2.0左右。od260/od280读数受测定所用溶液的ph值影响。同一个rna样品,假定在10mmtris,ph7.5溶液中测定出的od260/od280读数在1.8-2.1之间,在水溶液中所测定读数可能在1.5-1.9之间,但这并不代表rna不纯。浓度:去一定量的rna提取物,用rnase-free水稀释n倍,用rnase-free水将分光光度计调零,取稀释液进行od260测定,按照以下公式进行rna浓度的计算:终浓度(ng/μl)=od260×n(稀释倍数)×40.实施例3转录组测序分析1、rna-seq文库制备与测序质检合格的rna,用oligo(dt)的磁珠从总rna中富集mrna,在nebfragmentationbuffer中用二价阳离子将得到的mrna随机打断,以片段化的mrna为模版,随机寡核苷酸为引物,在m-mμlv逆转录酶体系中合成cdna第一条链,随后用rnaseh降解rna链,并在dnapolymerasei体系下,以dntps为原料合成cdna第二条链。纯化后的双链cdna经过末端修复、加a尾并连接测序接头,用ampurexpbeads筛选200bp左右的cdna,进行pcr扩增并再次使用ampurexpbeads纯化pcr产物,最终获得文库。文库构建完成后,先使用qubit2.0fluorometer进行初步定量,稀释文库至1.5ng/μl,随后使用agilent2100bioanalyzer对文库的insertsize进行检测,insertsize符合预期后,qrt-pcr对文库有效浓度进行准确定量(文库有效浓度高于2nm),以保证文库质量。库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行illuminahiseq测序。此部分委托北京诺禾致源科技股份有限公司完成。2、质量控制和比对分析2.1原始数据处理经过测序错误率检查、gc含量分布检查及原始数据过滤,获得后续分析使用的cleanreads,进行数据汇总。测序获得的原始数据中包含少量带有测序接头或测序质量较低的reads,为了保证数据分析的质量及可靠性,需要对原始数据进行过滤,过滤内容如下:(1)去除带接头的reads。(2)去除带有n碱基的reads。(3)去除测序质量低的reads。2.2比对分析采用star软件将cleanreads比对参考基因组序列。3、差异基因表达分析本研究采用fpkm来估算10个样品中所有的基因或转绿本的表达量。fpkm法同时考虑了测序深度和基因长度对reads计数的影响,是目前最为常用的基因表达水平估算方法。采用deseq2软件进行基因或转录本水平的表达差异显著性分析,将padj小于0.05作为差异显著性标准。基因差异表达分析的输入数据为基因表达水平分析中得到的readcount数据,分析主要分为三部分:(1)首先对readcount进行标准化(normalization);(2)然后根据模型进行假设检验概率(pvalue)的计算;(3)最后进行多重假设检验校正,得到fdr值(错误发现率)。4、go分析和kegg通路富集分析采用clusterprofiler软件对差异基因集进行go功能富集分析和kegg通路富集分析。通过对差异基因进行富集分析,找到差异基因与哪些生物学功能或通路显著性相关。5、结果本研究采用deseq2软件对正常组及nf1合并脊柱畸形组血液进行对比分析,筛选正常组与nf1合并脊柱畸形组中基因表达量padj值小于0.05的差异表达基因。最终共筛选出780个差异表达基因,包括319个表达上调的基因和461个表达下调的基因。go分析提示:这些差异基因主要来源于肌动蛋白细胞骨架等细胞成分;其分子功能主要为肌动蛋白结合、肌动蛋白丝结合等;主要参与蛋白质聚合、自然杀伤细胞介导免疫、肌动蛋白聚合或解聚、肌动蛋白丝长度的调节、肌动蛋白细胞骨架组织以及倒刺末端肌动蛋白丝加帽等生物过程。通路富集分析提示天然杀伤细胞介导的细胞毒性、补体途径以及移植物抗宿主疾病是差异基因参与的主要的信号传导通路。结合差异基因表达分析、差异表达基因的go、kegg富集分析以及文献检索数据对测序结果进行深度分析,最终发明人筛选出下调差异表达基因gpr56。gpr56可能是导致nf1合并脊柱畸形患者骨骼愈合缺陷的关键性分子。实施例4nf1合并脊柱畸形病外周血dna中gpr56基因表达情况1.实验材料获取自2018年1月至2018年4月于北京协和医院接受nf1合并脊柱畸形手术或检查的5例患者的外周血、5例相对正常的外周血和3例先天性脊柱侧凸的外周血样本。所有患者术前均未接受化疗或放疗,术前已取得患者的知情同意,并通过伦理委员会审核。样本处理:将edta抗凝全血按1:1比例与trizol混合,充分混匀后置于1.8ml细胞冻存管中,在液氮中迅速冷却30s后置于-80℃的冰箱保存。2.外周血rna的提取参照实施例2。3.逆转录合成cdna3.1第一链cdna合成试剂盒(revertaidpremiumreversetranscriptase)(thermoscientifictmep0733)3.2cdna第一链合成(1)在冰浴的nuclease-freepcr管中加入以下试剂:(2)轻轻混匀后离心3~5s,反应混合物在65℃温浴5min后,冰浴2min,然后离心3~5s。(3)将试管冰浴,再加入下列试剂:4.0μl5*rtbuffer0.5μlthermoscientificribolockrnaseinhibitor(20u)1.0μlrevertaidpremiumreversetranscriptase(200u)(4)轻轻混匀后离心3~5s(5)在pcr仪上按照下列条件进行反转录反应①25℃孵育10min②cdna合成50℃30min③终止反应85℃5min,处理后,置于冰上放置(6)将上述溶液-20℃保存。4.real-timepcr采用在线引物设计软件,基因序列参照ncbi:nm_001145770.2(gpr56),内参选gapdh,引物设计后由上海生工公司合成。具体引物序列如下:gpr56:seqidno.2gctacagccgaagaatgtgact;seqidno.3agcaggatgtttgggtttctc;扩增产物为125bp,扩增产物的核苷酸序列如seqidno.1所示。gapdh:seqidno.4tgggtgtgaaccatgagaagt;seqidno.5tgagtccttccacgataccaa。操作过程如下:反应体系如表1所示,用powergreenpcrmastermix进行扩增,实验操作按产品说明书进行。扩增程序为:95℃3min预反应;95℃3s,60℃30s进行45个循环的扩增反应。表1real-timepcr反应体系组分加入量2×mix10μl上游引物(10um)0.4μl下游引物(10um)0.4μl模板2μl加入灭菌蒸馏水至20μl将各样品cdna10倍稀释后取2μl作模板;所述pcr反应在abisteponeplus型荧光定量pcr仪中进行。5.实验结果根据qrt-pcr的相对定量公式,比较gpr56基因在nf1合并脊柱畸形病组和对照组中的表达水平。结果如图1显示,nf1合并脊柱畸形病组中gpr56基因的表达水平仅为对照组织的18.2%,以上结果验证了高通量转录组表达数据的整合分析gpr56基因在nf1合并脊柱畸形病低表达的结果。实施例5wb方法检测nf1合并脊柱畸形病中gpr56蛋白的表达1、实验材料同实施例42、各样本白细胞的分离与培养(1)外周血血5ml,肝素抗凝,1500r/min离心5min,取白细胞层。(2)加入1/3体积的3%明胶生理盐水溶液混匀,4℃静置20min。细胞成分分层,将上层混悬液移入另一试管中。直角离心机1750r/min离心10min。弃上清液,于底层细胞中加入蒸馏水3ml,充分振荡30s,加入等体积1.8%nacl3ml以除去混杂的红细胞。(3)1700r/min离心10min,弃上清液,底层细胞用pbs洗涤,1750r/min离心10min,弃上清液,加pbs配成1×106/ml细胞浓度备用。(4)细胞均置于含有10%胎牛血清(fbs)、100u/ml青霉素及100μg/ml链霉素、5%小牛血清的dmem培养基中于37℃、5%co2培养箱中传代培养。3、细胞总蛋白提取(1)将细胞从co2培养箱中取出,置于相差显微镜下观察,选择生长良好且铺满整个培养瓶瓶底的细胞用于蛋白质提取。提前准备预冷离心机至4℃。(2)准备冰盒备用,倒掉培养瓶中的旧培养基,用pbs洗涤培养瓶中残余培养基三次,倒置于滤纸上,尽量吸干水分后,迅速用细胞刮板将细胞刮下,加入1-2mlpbs2-3次,充分洗涤并收集pbs洗涤液置于离心管中并放入冰盒中。(3)将收集好的细胞1500rmp低温离心5min,弃上清。(4)每1x107个细胞加ripa裂解液(beyotime)1ml,置于冰上,并在在超声细胞破碎仪中超声3-5min。(5)放置10min后4℃下12000g离心10min,取上清,置于冰盒中备用。4、利用bca蛋白浓度测定试剂盒进行总蛋白定量采用康为世纪微量bca蛋白定量试剂盒(货号:cw2011),具体步骤见其说明书。5、sds-聚丙烯酰胺凝胶电泳(sds-page)(1)蛋白质样品变性:a)根据bca蛋白质浓度测定结果,每个凝胶加样孔中加入相同质量的总蛋白提取物。按照每1微升蛋白样品加入0.25微升蛋白上样缓冲液的比例,混合蛋白样品和蛋白上样缓冲液(5x)。b)100℃或沸水浴加热3-5分钟,以充分变性蛋白。c)冷却到室温后,直接上样到sds-page胶加样孔内即可。(2)胶板制备:采用bio-rad公司的微型垂直板电泳装置制备0.75mm厚的凝胶,照说明书安装好玻璃板后,先在小烧杯中配制5ml10%的分离胶,配方如下:表2分离胶配方组分用量30%丙烯酰胺溶液1.7mltris-hcl(1.5m,ph8.8)1.3ml10%sds0.05ml10%ap0.05mltemed0.002ml灭菌ddh2o补充至5ml混匀后立即灌胶,然后加lml蒸馏水覆盖,室温下放置约30min待胶聚合后,用蒸馏水洗2-3次,再用滤纸吸干。然后制备2ml5%的浓缩胶,配方如下:表3浓缩胶配方混匀后立即灌胶,插入样品梳,避免产生气泡,待胶凝固后,取出样品梳,后用蒸馏水和1x蛋白电泳缓冲液先后冲洗样品孔。6、上样及电泳将凝胶板装在电泳装置上,内槽中加满lx蛋白电泳缓冲液,外槽中lx蛋白电泳缓冲液应超过铂丝,按顺序上样。在末端泳道中加入蛋白质质量标准蛋白梯度。电泳时蓝色染料到达胶的底端处附近即可停止电泳。7、蛋白质印迹1)按照上述方法先进行sds-page凝胶电泳分离蛋白。2)预先用转印缓冲液浸泡nc膜、滤纸、海棉垫。sds-page结束后取出凝胶,去除浓缩胶,在tris/甘氨酸缓冲液中漂洗数秒,然后置于转印缓冲液中浸泡15-30min。打开电转印夹,每侧垫上一块专用的用转印缓冲液浸泡透的海棉垫,再各放一块转印液浸透的滤纸,滤纸与海棉垫大小相同或与nc膜,凝胶大小相同均可,将凝胶平放在阴极侧滤纸上,最后将nc膜平放在凝胶上,去除气泡,夹好电转印夹。在电泳槽加满电转印液,插入电转印夹,将电泳槽放入冰箱内(电转印液之前要放入冰箱内预冷),连接好电极,接通电流,转印夹的nc膜应对电泳槽的正极。3)封闭:用1xtbs漂洗一次。加入含5%的无脂奶粉tbs封闭缓冲液,置于振荡培养箱中进行封闭;4)一抗杂交:弃封闭液,加入用一抗稀释液稀释的一抗(anti-gpr56-antibody(ab77515))杂交溶液,置于4℃杂交过夜,第二天在振荡培养箱中进行杂交;5)回收一抗杂交液,用tbst洗膜3次;6)弃tbst,加入用封闭缓冲液稀释的二抗(goatanti-rabbitigg,hrpconjugated(cw0103))杂交溶液,置于振荡培养箱中进行杂交;7)弃二抗溶液,用tbst洗膜3次;8)ecl化学发光及图像采集和分析:按照高灵敏度化学发光检测试剂盒(康为世纪货号cw0049b),具体步骤参照说明书。9)以β-actin作为内参进行数据标准化,以正常对照组血白细胞中gpr56作为参照样本,计算各组血白细胞中gpr56蛋白的相对表达水平。五、实验结果结果如图2显示,nf1合并脊柱畸形患者中gpr56的表达水平显著低于正常组,gpr56的降低程度具有显著差异(p<0.05);在脊柱畸形患者(cs)中,gpr56蛋白的表达情况无统计学意义。实施例6gpr56对rsc96细胞增殖和周期的影响1.rnai干扰rsc96细胞中gpr56表达rsc96(大鼠雪旺细胞,是神经纤维瘤起源细胞)使用dmem培养基(gibco,货号12800017,添加nahco31.5g/l),90%,胎牛血清,10%;5%co2,37℃进行培养。取对数生长期的rsc96细胞,以5×104个/孔接种于24孔板中,过夜培养后,弃掉旧培养基,根据ribofecttmcp转染试剂说明书进行细胞转染,具体操作如下:①稀释sirna:用30μl1×ribofecttmcpbuffer稀释1.25μl20umsirna储存液,轻轻混匀。②混合液制备:加入3μlribofecttmcpreagent,轻轻吹打混匀,室温孵育0-15min。③将ribofecttmcp混合液加入到培养基中,轻轻混匀。将培养板置于37℃的co2培养箱中培养72h后。转染结束后收取细胞,通过westernblot检测sirna沉默效果。实验分为空白对照组con、阴性对照组nc、实验组sirna-1,sirna-2,sirna-3。westernblot结果显示,rsc96细胞在sirna浓度为50nm、转染72h后,转染组(sirna-1组,sirna-2组和sirna-3组)细胞中gpr56的表达水平与空白对照组和nc组相比明显降低,且降低效果明显,其中sirna-2和sirna-3的敲降效果有明显统计学意义(图3)。因此,在sirna浓度为50nm、转染72h下,rsc96细胞呈现出较高的转染效率,说明gpr56-sirna有效发挥作用,因sirna-2与sirna-3的干扰效果趋同,选取sirna-3进行后续相关实验。2.细胞增殖各组细胞转染24h,经0.25%的胰蛋白酶消化,用含10%小牛血清的dmem培养基配成单个细胞悬液,按每孔100μl,3×103个细胞接种于96孔板,并设5个复孔,37℃,5%co2培养箱中培养。分别于培养24、48、72、96h后,加入cck-8试剂,每孔10μl,经37℃5%co2培养箱中培养2h后,与450nm波长下检测每孔的吸光度值。3.细胞周期实验sirna-3转染的敲低gpr56的细胞株与对照组nc细胞分别铺于6孔板中,至细胞汇合度达85%~90%时,胰酶消化,完全培养基终止消化,2000rpm,5min,pbs2ml洗1次,收集细胞调整细胞浓度1×106/ml,取1ml单细胞悬液,离心,去除上清,加入70%冷乙醇500μl,4℃过夜固定,1000rpm,3min,弃上清,pbs清洗,离心,弃上清,每管加500μlpi/rnasea染色工作液,室温避光30-60min,流式细胞仪上机检测,记录激发波长488nm处红色荧光。结果显示,运用sirna敲低rsc96细胞中gpr56表达后,明显促进了rsc96细胞的增殖,具有统计学差异(图4);并改变rsc96细胞周期分布,影响了细胞周期进程,即缩短了处于g1期、s期的细胞,延长了处于g2期的细胞(图5)。实施例7rnai干扰成骨细胞mc3t3中gpr56表达后,对成骨相关基因的表达影响mc3t3使用rpmi-1640培养基(hyclone),10%胎牛血清,1%p/s;5%co2,37℃进行培养。取对数生长期的mc3t3细胞,以5×104个/孔接种于24孔板中,过夜培养后,弃掉旧培养基,根据ribofecttmcp转染试剂说明书进行细胞转染。转染结束后收取细胞,通过westernblot检测sirna沉默效果。实验分为空白对照组con、阴性对照组nc、实验组sirna-1、sirna-2、sirna-3。westernblot检测结果如图6所示,mc3t3细胞在sirna浓度为50nm、转染72h后,转染组细胞中gpr56的表达水平与空白对照组和nc组相比明显降低,且sirna-2和sirna-3的敲降效果有极显著差异。因此,在sirna浓度为50nm、转染72h下,mc3t3细胞呈现出较高的转染效率,说明gpr56-sirna有效发挥作用,因sirna-2与sirna-3的干扰效果趋同,选取sirna-3进行后续相关实验。进一步,通过westernblot检测gpr56表达下调后与成骨细胞分化相关标志物表达情况。所述成骨细胞分化相关的基因包括opn、ocn、alp等。结果如图7所示,转染组细胞中成骨分化的早期标志物alp和opn的表达水平与nc组及con组相比,降低趋势明显,且具有统计学差异;成骨细胞分化成熟的标志蛋白ocn的表达水平同样也呈下降趋势,但无统计学意义。在成骨细胞mc3t3细胞中敲降gpr56的表达水平后,不同程度上抑制了成骨细胞相关分化标志蛋白的表达,从而对成骨细胞的分化、细胞外基质成熟及矿化功能产生抑制。实施例8rnai干扰破骨细胞中gpr56表达后,对破骨相关基因的表达影响1.raw264.7破骨细胞的诱导分化取生长状态良好、细胞形态以圆形单核细胞占优势的raw264.7细胞,按2.5×104/cm2的密度接种到24孔板中,4-6h细胞贴壁后,每孔加入rankl,并调节成终浓度为50ng/ml,隔天半量换液,并保持rankl的终浓度不变,观察细胞形态特征变化,于培养的第1、3、5、7天按trap试剂盒说明书进行trap染色实验。2.trap染色细胞用pbs润洗两次,用4%多聚甲醛固定30min,然后用去离子水冲洗3次,根据trap试剂盒说明书配制染色液。具体操作如下:①足够的去离子水37℃预热备用;②0.5mlfastgarnetgbcbasesolution与0.5mlsodiumnitritesolution混匀30s后,静置2min;③取100ml烧杯混合加入如下溶液:37℃预热的去离子水45ml,gbc溶液1ml,naphtholas-biphosphqte0.5ml,acetatesolution2ml,酒石酸盐溶液1ml,将烧杯中的液体混匀后水浴加热至37℃;④将染液浸没细胞,37℃避光孵育1h;⑤1h后用离子水冲洗,再用hematoxylin复染2min。⑤碱性溶液冲洗数分钟;⑥晾干,倒置显微镜下观察:以trap染色阳性且含有3个细胞核以上的细胞作为破骨细胞进行计数。3.rnai干扰破骨细胞中gpr56表达取对数生长期的破骨细胞,以5×104个/孔接种于24孔板中,过夜培养后,弃掉旧培养基,根据ribofecttmcp转染试剂说明书进行细胞转染。转染结束后收取细胞,通过westernblot检测sirna沉默效果。实验分为空白对照组con、阴性对照组nc、实验组sirna-1,sirna-2,sirna-3。westernblot检测结果如图8所示,破骨细胞在sirna浓度为50nm、转染72h后,sirna-2组gpr56的表达水平与空白对照组和nc组相比降低较为明显,具有极显著差异,sirna-1和sirna-3的敲降效果不明显。因此,在sirna-2浓度为50nm、转染72h下,破骨细胞呈现出较高的转染效率,选取sirna-2进行后续相关实验。进一步,通过westernblot检测gpr56表达下调后破骨细胞相关标志物的表达情况。所述破骨细胞相关标志物包括nfatc1、c-fos等。结果如图9所示,转染组细胞中c-fos、nfatc1的蛋白表达水平与空白对照组及nc组相比均有不同程度降低,且降低程度均有极显著差异。在破骨细胞中敲降gpr56的表达水平后,进一步抑制了破骨细胞分化过程中重要的调节分子c-fos、nfatc1的表达,从而减少了有功能的多核破骨细胞的形成,抑制骨吸收功能。综上实验所述,敲降gpr56表达后,既影响了成骨细胞的形成,又影响了破骨细胞的吸收,但抑制破骨细胞的吸收比抑制成骨形成的程度更大(图7、图9),破坏成骨细胞与破骨细胞的平衡,进而导致骨疾病的发生。实施例9试剂盒的制作引物:特异扩增如seqidno.1所示的核苷酸序列的引物对如seqidno:2和seqidno:3所示;和特异扩增内参基因(gapdh)的引物对如seqidno:4和seqidno:5所示;还包括sybrgreen聚合酶链式反应体系,如pcr缓冲液、sybrgreen荧光染料、dntps。所述pcr缓冲液的成分为25mmkcl,2.5mmmgcl2,200mm(nh4)2so4。为方便使用,该试剂盒还可包含对照:正常人血液白细胞cdna。取受检者血液样本,使用常规方法(或使用特定的试剂盒)从血液样本的白细胞中提取rna,使用试剂盒中试剂,按照最佳反应体系与条件进行pcr反应,使用试剂盒中正常血液白细胞cdna作为real-timepcr定量检测中的对照cdna,检测受试者血白细胞中如seqidno.1所示的核苷酸序列相对血白细胞中表达量变化。此试剂盒的价值在于只需要外周血而不需要其他组织样本,通过最精简和特异的引物对检测基因的表达情况,不仅稳定,检测方便,且精确,大大提高诊断nf1合并脊柱畸形的敏感性和特异性,因此将此试剂盒投入实践,可以帮助指导诊断和更有效的个体化治疗。虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。序列表<110>中国医学科学院北京协和医院<120>与i型神经纤维瘤合并脊柱畸形病相关gpr56及其应用<130>p18024<160>5<170>siposequencelisting1.0<210>1<211>125<212>dna<213>genesequence<400>1gctacagccgaagaatgtgactctgcaatgtgtgttctgggttgaagaccccacattgag60cagcccggggcattggagcagtgctgggtgtgagaccgtcaggagagaaacccaaacatc120ctgct125<210>2<211>22<212>dna<213>人工序列(artificialsequence)<400>2gctacagccgaagaatgtgact22<210>3<211>21<212>dna<213>人工序列(artificialsequence)<400>3agcaggatgtttgggtttctc21<210>4<211>21<212>dna<213>人工序列(artificialsequence)<400>4tgggtgtgaaccatgagaagt21<210>5<211>21<212>dna<213>人工序列(artificialsequence)<400>5tgagtccttccacgataccaa21当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1