一种生物标志物、预测肾细胞癌的复发和死亡风险的方法与流程
本发明属于肾细胞癌监测技术,具体涉及一种可以用于预测肾细胞癌的复发和死亡风险的生物标志物、以及预测肾细胞癌的复发和死亡风险的的方法。
背景技术:
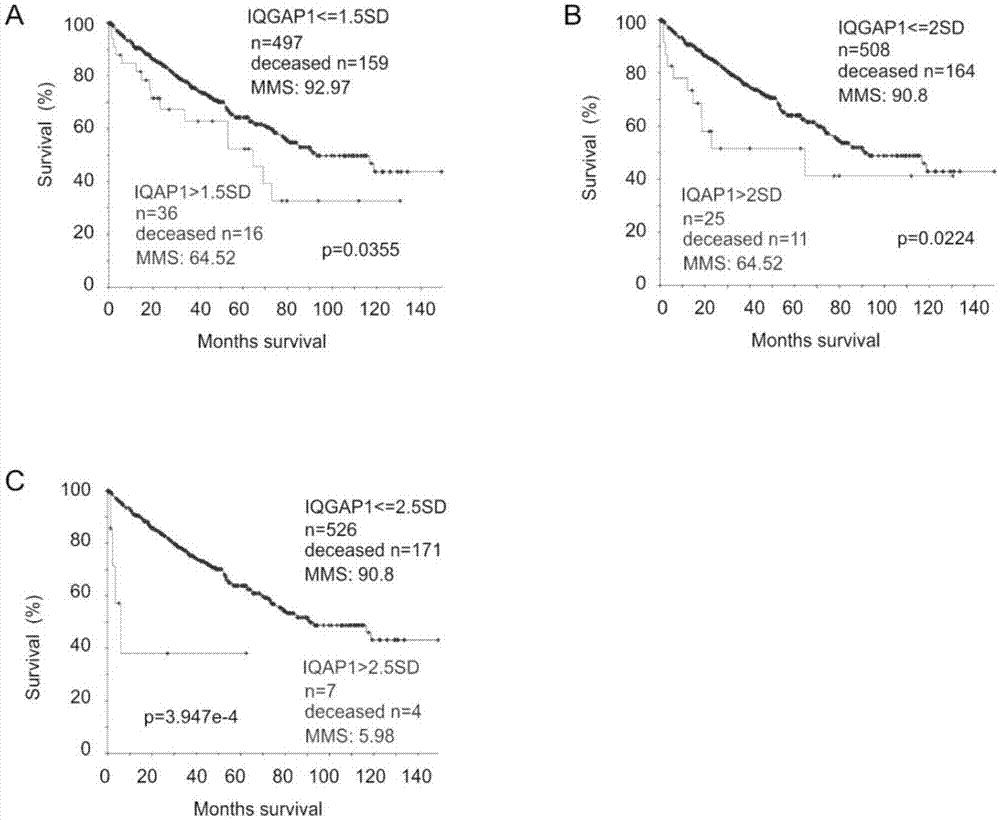
肾癌分别是男性中第九位和女性中第十四位常见的癌症。肾细胞癌(renalcellcarcinoma,rcc)占肾癌的85%,最常见的亚型为透明细胞癌ccrcc(80%),乳突细胞癌prcc(15%)和嫌色细胞癌chromophobercc、(5%)。其中,ccrcc是恶性程度最高的、造成最多人死亡的,对于原位ccrcc最主要的治疗方式是全部以及部分肾脏切除,这些患者中,30-40%术后都会发生复发和转移。目前,ccrcc转移癌还是无法治愈,面对这一状况,其中一个改善方法是对原位ccrcc患者根据复发、转移以及死亡的危险系数进行分类,为进行早期针对性治疗提供有力参考。虽然现在临床上已经使用肿瘤分期来预测预后情况,但是更完善的系统需要生物标志物的加入。目前,研究者们已经发现了一系列新的血清和尿液标志物,包括循环肿瘤细胞,不在细胞内的循环肿瘤dna,循环rna以及一些蛋白。这些标志物还需要大量的验证。最近,ccrcc的生物标志物领域被进行了系统的评估和升级。在被检查的28个标志物中,ccb是根据肿瘤分期分级调整后唯一一个独立标志物,更多有效的ccrcc标志物有待被开发。iqgap1属于支架蛋白家族(iqmotifgtpase-activatingscaffoldproteins,iqgaps)中的一员,iqgap1可以促进细胞外调节蛋白激酶(extracellularregulatedproteinkinases,erk)的活化,结合cdc42蛋白和rac1蛋白,稳定gtp的结合从而导致细胞骨架重组。研究还发现,iqgap1可以促进肿瘤发生,它在许多肿瘤中呈现过表达,其过表达水平与肿瘤细胞的侵袭和转移之间存在着密切的相关性,但是它在ccrcc中的作用还没有被报道过,因此iqgap1和其它相关的分子生物网络在有效地预测肾脏透明细胞癌患者的复发和死亡风险方面具有广阔的应用前景。技术实现要素:为了克服上述现有技术的缺陷,本发明所要解决的技术问题是:提供一种生物标志物及利用该生物标志物预测肾细胞癌复发和死亡风险的方法。为了解决上述技术问题,本发明采用的技术方案为:一种生物标志物,所述标志物用于预测肾细胞癌患者肾脏切除术后的复发或死亡风险,所述标志物至少包括以下两个基因:nsmaf、eif4g3、copb2、cdk20、amigo1、cables1、slc25a42、rab17、pgghg、pla2g6、spaca6、troap、pdxdc2p-npipb14p、slc25a27、ube2c、vamp1、parp10、lrrc61、top3b、cenpw、iffo1、ptprb、she、rspry1、sema3g、utp14c、akap6、nrp1、plpp3、ank3、amot、cavin2、ghr、ncoa4、rarb、sorbs2和gne。一种生物标志物,所述标志物用于预测肾细胞癌患者肾脏切除术后的复发或死亡风险,所述标志物为以下特征性基因组中的一组:sig2.5、sig0.83、sig0.83sub1、sig0.83sub2和sigcmbn;所述sig2.5包括以下基因:nsmaf、eif4g3、copb2、cdk20、amigo1、cables1、slc25a42和rab17;所述sig0.83包括以下基因:pgghg、pla2g6、spaca6、troap、pdxdc2p-npipb14p、slc25a27、ube2c、vamp1、parp10、lrrc61、top3b、cenpw、iffo1、ptprb、she、rspry1、sema3g、utp14c、akap6、nrp1、plpp3、ank3、amot、cavin2、ghr、ncoa4、rarb、sorbs2和gne;所述sig0.83sub1包括以下基因:spaca6、troap、cenpw和ncoa4;所述sig0.83sub2包括以下基因:pla2g6、spaca6、troap、pdxdc2p-npipb14p、ube2c、vamp1、top3b、cenpw、iffo1、ptprb、she、sema3g、utp14c、akap6、plpp3、ank3、amot、cavin2、ncoa4、sorbs2和gne;所述sigcmbn包括以下基因:nsmaf、eif4g3、copb2、cdk20、amigo1、cables1、slc25a42、rab17、pgghg、pla2g6、spaca6、troap、pdxdc2p-npipb14p、slc25a27、ube2c、vamp1、parp10、lrrc61、top3b、cenpw、iffo1、ptprb、she、rspry1、sema3g、utp14c、akap6、nrp1、plpp3、ank3、amot、cavin2、ghr、ncoa4、rarb、sorbs2和gne。进一步的,将所述生物标志物用于预测肾细胞癌患者肾脏切除术后的复发或死亡风险,所述肾细胞癌包括透明细胞癌和乳突细胞癌。进一步的,将所述生物标志物用于预测透明细胞癌患者肾脏切除术后的复发风险。进一步的,将所述生物标志物用于预测乳突细胞癌患者肾脏切除术后的复发风险。进一步的,将所述生物标志物用于预测乳突细胞癌患者肾脏切除术后的复发风险。进一步的,将所述生物标志物用于预测乳突细胞癌患者肾脏切除术后的死亡风险。进一步的,将所述生物标志物用于预测透明细胞癌患者肾脏切除术后的转移风险。进一步的,通过检测上述的生物标志物中特征性基因组的表达情况来预测肾细胞癌复发或死亡风险。进一步的,采用pcr、microarray、nanostring或rna测序的方法,以预测生物标志物中特征性基因组中基因的mrna高表达和mrna低表达。进一步的,对于生物标志物诊断阳性的标准,可以根据不同的人群和族群,以及使用不同的统计和计算机学习方法进行调整。进一步的,这些包含在生物标志物的基因也可以是这些基因的同种型或者基因的其它家族成员。进一步的,预测对象是人或哺乳动物。本发明的有益效果在于:本发明提供的生物标志物,充分利用iqgap1作为新型生物标志物的潜在价值,开发出多个组特征性基因组及子特征基因组,可以有效且高效地用于预测肾细胞癌的复发与死亡风险,进而利于更好的对病人进行管理,针对性的对部分复发或死亡风险高的病人进行新型辅助治疗或靶向治疗。附图说明图1所示为iqgap1上调与ccrcc总体生存率(os)间的关系;图2所示为iqgap1下调与ccrcc总体生存率间的关系;图3所示为两个特征性基因组的建立;图4所示为sig2.5组成基因的变化;图5所示为sig0.83组成基因的变化;图6所示为sig2.5和sig0.83和总体生存率间的关系;图7所示为sig2.5和ccrcc死亡风险的详细描述;图8所示为sig0.83和ccrcc死亡风险关联的详细描述;图9所示为sig0.83的组成基因对ccrcc死亡风险的预测;图10所示为sig2.5,sig0.83以及它们的亚特征性基因组与ccrcc复发间的关系;图11所示为sigcmbn对ccrcc和prcc的复发及死亡风险的预测;图12所示为sigcmbn和ccrcc死亡风险关联的详细描述;图13所示为sigcmbn中组成基因的表达情况和ccrcc死亡率间的关系;图14所示为sig2.5、sig0.83及sigcmbn和prcc的复发以及死亡风险间的关系;图15所示为预估ccrcc或prcc患者的五年生存率的临床列线图;图16所示为sig2.5和sig0.83的临床列线图;图17所示为sig2.5、sig0.83和sigcmbn预测ccrcc和prcc的五年生存率;图18所示为sig0.83和ccb对ccrcc死亡风险的预测。具体实施方式为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式并配合附图予以说明。本发明从tcgaprovisionalccrcc的数据库(n=533)中,挑选出了56个相关iqgap1升高和2726个相关iqgap1降低的差异性表达基因(degs)、利用elastic-net回归模型,研究了上述degs对ccrcc死亡率的影响,从56个相关iqgap1升高的degs中建立起了特征性基因组sig2.5,sig2.5包含nsmaf、eif4g3、copb2、cdk20、amigo1、cables1、slc25a42和rab17,8个基因;从2726个相关iqgap1降低的degs中建立了特征性基因组sig0.83,sig0.83包含pgghg、pla2g6、spaca6、troap、pdxdc2p-npipb14p、slc25a27、ube2c、vamp1、parp10、lrrc61、top3b、cenpw、iffo1、ptprb、she、rspry1、sema3g、utp14c、akap6、nrp1、plpp3、ank3、amot、cavin2、ghr、ncoa4、rarb、sorbs2、和gne,29个基因。从sig0.83得到两个子特征性基因组sig0.83sub1和sig0.83sub2,sig0.83sub1包括spaca、troap、cenpw和ncoa4,4个基因;sig0.83sub2包括pla2g6、spaca6、troap、pdxdc2p-npipb14p、ube2c、vamp1、top3b、cenpw、iffo1、ptprb、she、sema3g、utp14c、akap6、plpp3、ank3、amot、cavin2、ncoa4、sorbs2和gne,21个基因。合并sig2.5和sig0.83得到sigcmbn。sig2.5、sig0.83、sig0.83sub1、sig0.83sub2和sigcmbn都能够有效的预测不同亚型的肾细胞癌患者的复发和死亡风险,这些亚型包括透明细胞癌和乳突细胞癌。sig2.5预测ccrcc的死亡风险的敏感性/特异性/阳性预测率(positivepredictivevalue,ppv)/mms/p为87.4%/93.6%/58.5%/31.3/4.55e-15;sig0.83预测ccrc的死亡风险的敏感性/特异性/ppv/mms/p值为89.7%/90.2%/71.8%/31.1/0;sig0.83sub1预测ccrcc的死亡风险的敏感性/特异性/ppv/mms/p值为62.3%/83.8%/65.3%/36.5/0;sig0.83sub2预测ccrcc的死亡风险的敏感性/特异性/ppv/mms/p值为62.3%/83.8%/65.3%/36.5/0;sigcmbnccrcc预测ccrcc的死亡风险的敏感性/特异性/ppv/mms/p值为90.3%/89.1%71.2%/31.1/0。在调整诊断时病人的年龄和肿瘤分级分期后,sig2.5、sig0.83和sigcmbn可以作为预测ccrcc死亡风险的独立危险因子;调整诊断时的病人年龄和肿瘤分级分期后,sig2.5的8个基因中的6个基因和sig0.83的29个组成基因也可以作为预测ccrcc死亡风险的独立危险因素,上述基因都和ccrcc的转移呈显著相关。用tcgaprcc数据库(n=291)来验证,sig2.5、sig0.83和sigcmb均与os的下降显著相关。sig2.5鉴别出肿瘤死亡风险的tauc81.7%/12.4m(month,月),81.3%/20m,80.8%.30.4m,and76.6/56.8m来排除高风险死亡的肿瘤;sig0.83鉴别出肿瘤死亡风险的tauc87.5%/12.4m(month,月),87.8%/20m,87.8%.30.4m,and75.9/56.8m排除prcc死亡风险;sigcmbn预估prcc死亡风险的敏感性/特异性/ppv/mms/p值为47.7%/93.1%/55.3%/20.5%/0,同时它高效预测prcc复发风险的敏感性/特异性/ppv/无疾病中位生存期(medianmonthsdisease-free,mmdf)/p值为41.5%/94.4/64.7%/9.72/0。实施例1:1实施样品与方法1.1病例样品选择cbioportal数据库中的tcgaprovisionalccrcc数据库中533例原位癌病人,所有的肿瘤都被外科切除并且rna测序得到了rna的表达情况,以及140个月的随访信息和部分临床数据。选择tcgaprovisionalprcc数据库中291例肿瘤/病人经过了rna测序,以及随访信息和部分临床数据。tcga中ccrcc病人的人口统计学资料如表1所示,其中,q为四分位数(quartile),na为无可用数据(notavailable),tartmoltherapy为靶向分子治疗:表11.2基因富集分析用r语言里的gage(49)和reactome(50)语言包,分析了富集的基因集合和通路。数据库使用了kegg(kyotoencyclopediaofgenesandgenomes)和go(geneontology)数据库。1.3回归分析逻辑回归使用了r语言进行,cox比例风险模型(coxph)回归分析使用了r语言中的survival语言包,对是否符合比例风险(ph)假定也进行了验证。1.4多特征性基因组的建立tcgaprovisionalccrcc数据库中随着iqgap1升高(n=56)和下调(n=2726)的基因被用来建立了特征性表达基因。特征性表达信息,随访时间,复发情况,总体生存率,肿瘤分级分期和远期转移情况都从数据库当做提取了出来。根据对总体生存率的影响,采用r语言中的glmnet语言包对degs进行了elastic-net回归分析(十倍交叉验证)。混合参数α采用了0.2,0.5和0.8。当α=0,elastic-net呈现为ridge回归,相关预测因子的回归系数被缩小,不能选出协变量;当α=1,它表现为lasso回归,会在一组变量中只选出一个协变量。这些都会降低这些标志物的预测潜能。所以为了选出一组高相关的变量,我们采用了三个α值,0.2,0.5和0.8,进行了三次elastic-net回归分析。1.5病人/肿瘤分配的标志物得分所有的组成基因和生存率降低之间的关系都用r语言中的survival语言包进行了单变量cox比例风险回归分析。在都符合ph假定情况下,得到了各个degs的cox相关系数(coxcoefficients)。相关系数求和得到了每个病人的基因组分数。1.6割点分数(cutpoint)的估计判断肿瘤高风险死亡的割点分数是使用r语言中的maxstat语言包进行最大选择log-rank检验(maximallyselectedrankstatistics)得到的。从tcgaccrcc和prcc数据库中得到了每个组成基因的rna表达情况后,依照此方法得到了它们的割点分数。1.7列线图(nomogram)的建立用r语言中的rms语言包建立起了各个特征性基因组为基础的列线图,用于预测ccrcc或者prcc病人的生存率。所有列线图都用200次自展法(bootstrap)验证过了,也经过了矫正。c-index用somers’d(dxy)=2x(c-0.5)算出。1.8统计分析fisher’s精确检验(fisher’sexacttest)由graphpadprism5完成。kaplan-meier生存曲线和log-rank检验由r语言中的survival语言包和cbioportal完成。单变量和多变量cox回归分析由r语言的survival语言包完成。时间依赖受试者操作曲线(troc)由r语言中的timeroc语言包完成。p值<0.05被当做有显著统计学差异。2实施过程与结果2.1iqgap1水平和ccrcc总体生存率下降的双向关系表1中含有533例手术切除的原位ccrcc患者,其mrna表达情况已用rna测序得到,这些病例被随访了140个月,参考的群体采用数据库中没有染色体数目异常的肿瘤的相关基因或者整个肿瘤群体。和其他癌症类型一致的是,iqgap1比参考群体平均值上调1.5个标准差(1.5sd),2sd以及2.5sd和ccrcc的总体生存率os下降呈显著相关,并且相关强度随着iqgap1的升高而升高(如图1所示)。使用了r语言中的maxstat语言包对iqgap1的mrna表达水平进行了最大选择rank检验(maximallyselectedrankstatistics),根据ccrcc的死亡风险高低对其预估出了一个割点分数(如图2所示),iqgap1表达为-0.83sd,-1sd以及-1.5sd都和ccrcc总体生存率下降相关(如图2b-d所示),其中割点分数-0.83sd显示了与总体生存率下降最好的相关关系(如图1和图2所示),可以看出在单一的癌症类型–ccrcc中,其不良预后和iqgap1的水平呈双向关系。2.2iqgap1表达变化的相关基因用图3a所示的方法对tcga数据库中与iqgap1上调2.5sd和下调0.83sd相关的转录组进行了分析。在iqgap1表达上调2.5sd的7个肿瘤和无上调的526个肿瘤中,分析出了56个差异化表达基因(degs)(q<0.05),在iqgap1下调0.83sd相关的转录组中得到了2726个degs(q<0.001,差异倍数≥1.5)。其中,这56个degs主要影响tnf通路,2726个degs则表达于多个通路,包括柠檬酸(tca)循环、呼吸相关电子传送、翻译起始、vegf通路和其他通路;然后用kegg数据库(r语言中的gage语言包)对这2726个degs进行了基因富集分析。其中上升的基因集涉及到氧化磷酸化和核糖体,支持其影响线粒体中的呼吸电子传导以及蛋白质的合成。另外,下降的基因集除了在局部粘附(focaladhesion),肌动蛋白(actin)细胞骨架调节和钙信号传导中都起作用,还影响一系列神经退行性疾病和心肌病。2.3预测ccrcc肾脏切除后总体生存率的特征性基因组sig2.5,sig0.83和sigcmbn的建立分别从56个degs和2726个degs中建立了两个与ccrcc总体生存率相关的两个特征性基因组。采用了等价的交叉验证法。用-1sd和+2.5sd当做截断点把56个degs分为了上调和下调两组,用-1sd和2sd当做截断点把2726个degs分为了两组(如图3a所示);然后用elastic-net回归分析(r语言的glmnet语言包)分析了56个degs和2726个degs对总体生存率的影响(如图3a所示)所示。elastic-net分析用0.2,0.5和0.8三组混合参数α,并且对每一组进行了十倍的交叉验证。三组设置里的核心基因以及0.2组里的其他基因组成了最后的特征性基因组,包括来自56个degs的8个基因的sig2.5和来自2726个degs的29个基因的sig0.83(如图4和图5所示);用-1sd和2.5sd作为每个基因的截断点,sig2.5和ccrcc总体生存率os下降(p=4.32e-6)以及无疾病生存期dfs下降(p=1.65e-4)显著相关(如图3b和图3c所示);用-1sd和2sd作为截断点,sig0.83与总体生存率os下降(如图3d所示,p=3.29e-11)以及无疾病生存期dfs(如图3e所示,p=2.44e-8)呈更显著的相关。2.4sig2.5和sig0.83的优化并应用于预测ccrcc的死亡风险用最大选择log-rank检验(如图2所示)对所有组成基因预测ccrcc死亡风险的割点分数进行了分析,所有肿瘤都由此被赋予了各个组成基因的二进制密码。对于上升的degs,若其表达水平>割点分数,肿瘤就被定义为“1”,否则肿瘤被定义为“0”。对于下降的degs,肿瘤被定义了相反的二进制密码。在所有degs都满足比例风险(ph,proportionhazards)假定的情况下,用割点分数当做截断点,对它们进行了单变量cox比例风险模型分析,每个肿瘤的sig2.5和sig0.83累积得分为∑(fi)n,其中fi:每个基因的系数估计值,sig2.5的n=8,sig0.83的n=29。这两个分数预测ccrcc死亡风险的能力用时间依赖曲线下面积表示(tauc,time-dependentareaundercurve):sig2.5的tauc范围为70.4%/13.8m到72.6%/70.6m,sig0.83的tauc范围77.1%/13.8m到79.6%/70.6m(如图6所示),然后将两个割点分数当做截断点,即sig2.5cut和sig0.83cut,给两个基因组赋予了二进制代码,sig2.5cut和sig0.83cut和ccrcc总体生存率os下降显著相关(如图6b和图6c所示)。相比于用sd当做截断点(如图3b和图3d所示),sig2.5cut以及sig0.83cut和ccrcc总体生存率下降的相关更强,所用依据是它们的特异性/ppv/mms/p值:sig2.5sd的为63.1%/43,3%/62.8/4.23e-6(如图3b所示),相比之下sig2.5cut的为93.4%/67.6%/31.3/4.55e-15(如图6b所示);sig0.83sd的为51.4%/44.6%/62.8/3.29e-11(如图3d所示),相比之下sig0.83cut的为88.8%/71.2%/31.1/0(如图6c所示)当用第一四分位数(q1),中位数(median)和第三四分位数(q3)分别当做截断点,sig2.5和sig0.83都可以很好的预测ccrcc的死亡风险(如图7a-c和图8a-c)所示;当把割点分数、q1、中位数和q3放在一起用,sig2.5和sig0.83的敏感性/特异性/ppv/mms/p值可以达到最佳:分别为87.4%/93.6%/67.6%/31.3/4.55e-15(如图7d所示)和89.7%/90.2%/71.8%/31.1/0(如图8d所示);在sig2.5中,除了eif4g3,所有组成基因都是prcc死亡的高风险因素(如表2所示),sig2.5的累积分数预计prcc死亡风险的tauc为81.7%/12.4m,81.3%/20m,80.8%.30.4m和76.6/56.8m,sig2.5cut和prcc总体生存率下降也显著相关(如图14a所示);sig0.83cut和prcc总体生存率的下降显著相关(如图14b所示),预测prcc死亡的tauc为87.5%/12.4m(month),87.8%/20m,87.8%.30.4m和75.9/56.8m;表2genescoefhr95%cipvaluensmaf1.18493.271.797-5.9510.000105***eif4g30.78162.1850.858-5.5640.101copb21.22473.4031.737-6.6680.000358***cdk201.63015.1052.677-9.7347.45e-7***amigo10.95772.6061.316-5.1610.00602**cables11.29473.651.744-7.6390.000591***slc25a420.79572.2161.215-4.0410.00943**rab171.62765.0912.717-9.543.78e-7***实施例2:所用实施样品、方法与实施例1相同;将sig0.83sub1和sig0.83sub2用于预测crcc死亡风险:sig0.83中的spaca6、troap、cenpw和ncoa4的hr较高,将其分出作为sig0.83的第一个亚基因组sig0.83sub1,同时p值>2.79e-9的degs从剩余基因里被剔除,留下21个基因组成sig0.83sub2;sig0.83sub1、sig0.83sub2和sig0.83cut预测ccrcc死亡风险的敏感性/特异性/ppv/mms/p值分别为41.1%/93.6%/75.8%/27.2/0(如图6d所示),62.3%/83.8%/65.3%/36.5/0(如图6e所示)和50.9%/90.2%/71.8%/31.1/0(如图6c所示)。sig0.83sub1和sig0.83sub2鉴别ccrcc死亡风险的tauc在四个时间框架内分别大于70%和76%(如图6a所示)。实施例3:所用实施样品、方法与实施例1相同;将sigcmbn用于预测ccrcc死亡风险:sigcmbn预测ccrcc高死亡风险的tauc为78.1%/13.8m,78.7%/31m,77.8%/48.9m和81.3%/70.6m,高于sig2.5或sig0.83(如图11a和图6a所示);sigcmbn的割点分数(如图11b所示),q1,中位数,平均数和q3(如图12所示)都和总体生存率下降相关,用q1,中位数,q3和割点分数一起预测死亡风险的话,敏感性/特异性/ppv/mms/p值可以达到最好的90.3%/89.1%/71.2%/31.1/0(如图11c所示);sigcmbn的37个组成基因中,有25个基因的割点分数都各自和总体生存率下降相关(p≤0.092),其累积分数预测死亡风险的tauc为76%/23.2m,82.1%/36.3m,86.5%/45.5m和80.7%/58.9m(如图13a所示),这25个基因组成的基因组的极端点也和总体生存率下降显著相关;sigcmbn在预测prcc死亡风险上其敏感性/特异性/ppv/mms/p值为47.7%/93.1%/55.3%/20.5/0(如图11e所示),和生存曲线一致的是(如图11e所示),根据tauc的结果(如图11f所示),sigcmbn鉴别出早期死亡要更高效。同时sigcmbn也可以很好的预测prcc的复发风险,其敏感性/特异性/ppv/mmdf(medianmonthsdisease-free,无疾病中位生存期)/p值为41.5%/94.4/64.7%/9.72/0(如图14c所示)。实施例4:使用sig2.5,sig0.83和sigcmbn建立列线图(nomograms)预估肿瘤的生存率;用r语言中的rms语言包建立了列线图来预估五年生存率。sig2.5,sig0.83和sigcmbn各自累计分数,诊断年龄和肿瘤分级分期被用于制作相应的列线图(如图15a和图16所示),可以看出用sigcmcn分数得到的列线图(图15a)和用sig0.83分数得到的列线图(如图16b所示)优于用sig2.5得到的列线图(如图16a所示);用一致性指数和矫正检验了这些列线图的区别度。经过两次自展法验证,用sig2.5分数,sig0.83分数和sigcmbn分数制作的列线图的c-index分别为0.78,0.83和0.81,基于校正后的曲线(如图17a-c所示),用sig2.5分数,sig0.83分数和sigcmbn分数制作的列线图都可以预测五年生存率。其中相比另外两条校正后曲线,用sigcmbn分数制作的列线图(如图17c所示),和理想预测曲线更为接近(如图17a和图17b所示);根据prcc的sigcmbn分数和诊断年龄也建立起了相应的列线图(如图15b所示),其鉴别prcc死亡风险的c-index为0.87,校正后曲线显示其预测生存率优于sigcmbn列线图预测ccrcc生存率。实施例5:将sig2.5、sig0.83和sigcmbn用于分析肿瘤转移、分级分期sig2.5(比值比,oddsratio/or3.48,95%ci1.96-6.11,p=1.64e-5),sig0.83(or4.51,95%ci2.72-7.51,p=5.66e-9)和sigcmbn(or4.98,95%ci3.02-8.29,p=5.66e-10)都和ccrcc转移显著相关(如表3所示);sig2.5,sig8.3和sigcmbn以及它们的组成基因也和肿瘤的高分级分期相关性如表3所示,为sig2.5,sig0.83,sigcmbn和其组成基因与ccrcc不良预后之间相关性;表3实验例1:将sig2.5、sig0.83、sig0.83sub1、sig0.83sub2和sigcmbn与现有的ccrcc生物标志物作比较现有ccrcc的生物标志物ccb,使用了tcgaprovisionalccrcc数据库的一个子集,一共350个病例,ccb基因组由24个基因组成,其上调和ccrcc恶性程度更高的亚型相关。将现有ccrcc生物标志物与sig0.83中上调的13个基因(sig0.83ups)进行了比较,这两组基因没有重叠,其中ccb的基因为aldh1a2、ap4b1、b3gnt8、bcl2l12、reep2、cdh3、cyb5r2、flj23867、galnt10、igf2bp2、kcnk6、kcnn4、matn4、fam133b、ube2f、npm3、saa4。slpi、sytl1、tpm4、ttll3、ung、usp4和znf292:结果发现,在高于平均值平均值1sd到3sd的范围内,两个基因组都和os下降相关,其中2.5sd的相关是最显著的;相比较而言sig0.83up优于ccb(如图18a和18b所示);在1sd到3sd这个范围内,ccb在1.5sd-2.5sd可以预测疾病复发,而sig0.83up在整个范围内都可以预测ccrcc的复发;预测无疾病生存期dfs方面,sig2.5,sig0.83,sig0.83sub1,sig0.83sub2和sigcmbn都优于ccb。实验例2:分析sig2.5和sig0.83是否为ccrcc死亡的独立危险因子,结果如表4所示:表4表4为sig2.5,sig0.83,sigcmbn和其组成基因与ccrcc总体生存率之间关系的单变量以及多变量cox模型分析结果,从表4中可以看出,sig2.5(危险系数hr2.23;95%ci1.54-3.23,p=2.15e-5),sig0.83(hr3.21;95%ci2.31-4.45,p=3.16e-12)和sigcmbn(hr3.69;95%ci2.66-5.11,p=4.33e-15)都可以作为预测ccrcc死亡的独立危险因子;sig2.5的8个组成基因中的6个基因和sig0.83的29个组成基因都是独立风险因子,其中有些基因的危险系数还很高。这进一步证明了sig2.5,sig0.83,sigcmbn能够准确地预测ccrcc的死亡风险。综上所述,本发明提供的一种生物标志物、预测肾细胞癌的复发和死亡风险的方法,通过利用iqgap1及相关分子生物网络,预测肾细胞癌患者的复发和死亡风险,ccrcc中的不良预后和iqgap1的水平呈双向关系,利用iqgap1相关的degs,建立了sig2.5和sig0.83两个特征性基因组,通过对sig2.5和sig0.83组合得到了特征性基因组sigcmbnsig,三者能够有效地预测ccrcc切除肾脏以后的复发率和总体生存率,预测ccrcc的死亡风险,同时通过组合sig0.83中不同的基因得到两个子特征性基因组,可以更好的预测ccrcc的死亡风险;本发明提供的生物标志物,充分利用iqgap1作为新型生物标志物的潜在价值,开发出多个组特征性基因组及子特征基因组,可以有效且高效地用于预测肾细胞癌的复发与死亡风险,进而利于更好的对病人进行管理,针对性的对部分复发或死亡风险高的病人进行新型辅助治疗或靶向治疗。以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的
技术领域:
,均同理包括在本发明的专利保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1