法庭科学用氟氯唑仑及其制备方法和应用与流程
本发明涉及法庭科学毒物
技术领域:
。具体地说是法庭科学用氟氯唑仑及其制备方法和应用。
背景技术:
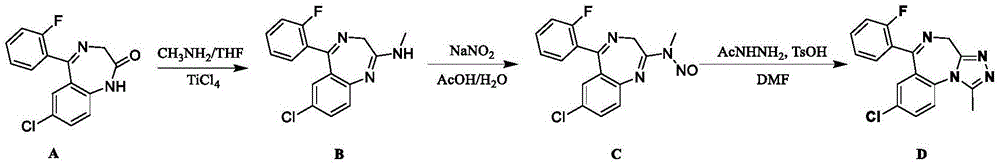
:2017年以来,全国各地相继出现许多麻醉强奸案件,犯罪嫌疑人网购标有“弥漫之夜”、“迷恋”、“猎艳”、“情愫”等字样的成分不明液体,掺入酒水、饮料中诱骗女子喝下,女子喝下含药物成分的酒水、饮料后很快失去知觉与抵抗能力。各地公安机关在对收缴的药物及受害人生物检材血液、尿液等采用免疫胶体金试剂条筛查时,苯二氮卓类试剂条呈阳性,但采用仪器进行检验时,大多却无法检出《麻醉药品和精神药品目录》、《非药用类麻醉药品和精神药品》中的药物成分。犯罪嫌疑人往往狡辩未给女方下药、女方自愿发生性关系等。同时,也出现了采用该类药物实施抢劫、凶杀的案件。经过采用高分辨q-tof/ms液质联用仪、气质联用仪及核磁共振分析,我们确证了该类检材中往往含有未被nist库收录的、许多公安机关检验鉴定机构尚不熟悉的苯二氮卓类麻醉策划药:8-氯-6-(2-氟苯基)-1-甲基-4h-[1,2,4]三唑并[1,4]苯并二氮卓(中文名:氟氯唑仑)。该物质属不法分子最新合成的苯二氮卓类策划药,国内外均缺乏标准物质,公安机关往往无法出具检验报告、有效打击犯罪嫌疑人。因此,网络流行苯二氮卓类策划药氟氯唑仑的研制与应用在提高公安机关理化检验能力、提供侦查破案诉讼证据、有效打击犯罪等方面具有十分重要的意义。技术实现要素:为此,本发明所要解决的技术问题在于提供一种法庭科学用氟氯唑仑及其制备方法和应用。采用该方法制备的氟氯唑仑,纯度高达99%以上,可直接用作公安机关检验鉴定工作的对照品。为解决上述技术问题,本发明提供如下技术方案:法庭科学用氟氯唑仑,如下式所示:法庭科学用氟氯唑仑的制备方法,包括如下步骤:(1)由化合物a合成化合物b;(2)由化合物b合成化合物c;(3)由化合物c合成氟氯唑仑化合物d;所述化合物a如下式所示:所述化合物b如下式所示:所述化合物c如下式所示:上述法庭科学用氟氯唑仑的制备方法,在步骤(1)中,(1-1)称取一定质量的化合物a加到三口瓶中,再加入甲胺的四氢呋喃溶液,搅拌,氮气保护,低温下,开始缓慢滴加四氯化钛,并控制反应温度,20分钟滴完;(1-2)滴完后,升温进行反应;tlc监控反应,原料消失,停止反应;(1-3)反应结束后,向反应液中滴加水猝灭反应,再加入水和乙酸乙酯,萃取分液,保留上层有机相;(1-4)上层有机相用饱和食盐水洗涤,然后用无水硫酸钠干燥;(1-5)过滤,并用乙酸乙酯冲洗滤饼,合并滤液和洗液,减压浓缩干,得到化合物b。上述法庭科学用氟氯唑仑的制备方法,在步骤(1)中:(1-1)称取0.0139mol的化合物a加到250ml三口瓶中,再加入100ml甲胺的四氢呋喃溶液,搅拌,氮气保护,冰盐浴降温至内温为-5℃,开始缓慢滴加0.0139mol四氯化钛,控制反应温度不高于5℃,并在20分钟滴完;所述甲胺的四氢呋喃溶液中,甲胺的物质的量浓度为2mol/l;(1-2)滴完后,升温至20℃-25℃反应4小时,再升温至40℃-45℃反应1小时;反应过程中,采用tlc监控反应,原料消失,停止反应;(1-3)反应结束后,向反应液中滴加50ml水猝灭反应,再加入50ml水和50ml乙酸乙酯,萃取分液,保留上层有机相;(1-4)上层有机相用饱和食盐水洗涤一次,然后用无水硫酸钠干燥;(1-5)过滤,并用乙酸乙酯冲洗滤饼两次,乙酸乙酯每次用量为20ml,合并滤液和洗液,减压浓缩干,得到化合物b。上述法庭科学用氟氯唑仑的制备方法,在步骤(2)中:(2-1)化合物b加到三口瓶中,依次加入乙酸和水,搅拌,降至低温,开始缓慢滴加亚硝酸钠水溶液;(2-2)滴完后,升温进行反应;(2-3)反应结束后,向反应液中加入乙酸乙酯,再滴加饱和碳酸钠水溶液调节ph值;(2-4)然后分液,保留有机相,水相再用乙酸乙酯萃取,合并有机相,然后用饱和食盐水洗涤有机相,用无水硫酸钠干燥;(2-5)过滤,并用乙酸乙酯冲洗滤饼,合并滤液和洗液,减压浓缩干,层析柱纯化,得到化合物c。在步骤(2)中:(2-1)0.0136mol化合物b加到150ml三口瓶中,依次加入25ml乙酸和5ml水,搅拌,冰水浴降温至内温3℃,开始缓慢滴加7.2ml亚硝酸钠水溶液,亚硝酸钠的物质的量浓度为4.72mol/l,15分钟滴完;(2-2)滴完后,升温至25℃反应3小时;(2-3)反应结束后,向反应液中加入40ml乙酸乙酯,再滴加饱和碳酸钠水溶液调节ph值至8;(2-4)然后分液,保留有机相,水相再用40ml乙酸乙酯萃取一次,合并有机相,然后用50ml饱和食盐水洗涤有机相,用无水硫酸钠干燥;(2-5)过滤,并用乙酸乙酯冲洗滤饼2次,乙酸乙酯每次为15ml,合并滤液和洗液,减压浓缩干,层析柱纯化,淋洗剂为石油醚pe和乙酸乙酯ea的混合物,pe与ea的体积比为20:1,收集产品,得到化合物c。上述法庭科学用氟氯唑仑的制备方法,在步骤(3)中:(3-1)化合物c加到单口瓶中,依次加入n,n-二甲基甲酰胺、对甲苯磺酸和乙酰肼,加热反应,tlc监控反应;(3-2)停止反应,降温至室温,加入饱和碳酸氢钠水溶液和二氯甲烷,萃取分液,保留有机相,水相再用二氯甲烷萃取,合并有机相;然后用饱和食盐水洗涤有机相,用无水硫酸钠干燥;(3-3)过滤,并用二氯甲烷冲洗滤饼,合并滤液和洗液,减压浓缩干,层析柱纯化,得到化合物d粗品;(3-4)化合物d粗品溶于二氯甲烷,再缓缓滴加异丙醚,有固体析出,过滤,并用异丙醚冲洗滤饼,取滤饼,抽干,得到化合物d。上述法庭科学用氟氯唑仑的制备方法,在步骤(3)中:(3-1)4.544mmol化合物c加到100ml单口瓶中,依次加入20mln,n-二甲基甲酰胺dmf、2.499mmol对甲苯磺酸和5.453mmol乙酰肼,加热至120℃反应4小时,tlc监控反应;(3-2)停止反应,降温至室温,加入40ml饱和碳酸氢钠水溶液和30ml二氯甲烷,萃取分液,保留有机相,水相再用二氯甲烷萃取2次,二氯甲烷每次用量为20ml,合并有机相;然后用50ml饱和食盐水洗涤有机相,用无水硫酸钠干燥;(3-3)过滤,并用二氯甲烷冲洗滤饼2次,二氯甲烷每次用量为10ml,合并滤液和洗液,减压浓缩干,层析柱纯化,淋洗剂为二氯甲烷和甲醇的混合液,二氯甲烷和甲醇的体积比为50:1,收集纯度高的产品点,得到化合物d粗品;(3-4)化合物d粗品溶于3ml二氯甲烷,再缓慢滴加15ml异丙醚,有固体析出,过滤,并用异丙醚冲洗滤饼2次,异丙醚每次为4ml,取滤饼,抽干,得到化合物d。法庭科学用氟氯唑仑的应用,上述氟氯唑仑可用于公安机关检验鉴定工作的标准物质。本发明的技术方案取得了如下有益的技术效果:1.优化合成法庭科学用氟氯唑仑的最佳合成路线。2.分离纯化目标物法庭科学用氟氯唑仑,使得法庭科学用氟氯唑仑纯度达到99%以上。3.所制备的法庭科学用氟氯唑仑可直接用作公安机关检验鉴定工作的对照品。附图说明图1本发明法庭科学用氟氯唑仑制备方法的合成路线图;图2本发明法庭科学用氟氯唑仑的分子式;图3本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的核磁共振氢谱图(400.13mhz,cdcl3,297k);图4本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的核磁共振碳谱图(100.6mhz,cdcl3,297k);图5本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的1h/13chmbc谱图(cdcl3,297k);图6本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的1h/13chmqc谱图(cdcl3,297k);图7本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的1h/1hcoesy谱图(cdcl3,297k);图8发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的核磁共振氟谱图(376.5mhz,cdcl3,297k);图9a本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的lc-q-tof-ms的tic谱图;图9b本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的lc-q-tof-ms的eic谱图图10a本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的lc-q-tof-ms在正离子模式下一级质谱图;图10b本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的lc-q-tof-ms在正离子模式下的二级质谱图;图11本发明法庭科学用氟氯唑仑制备方法所得到的氟氯唑仑的hplc谱图。具体实施方式一、法庭科学用氟氯唑仑的如下式所示:二、法庭科学用氟氯唑仑的制备方法,包括如下步骤:(1)由化合物a合成化合物b:(1-1)称取0.0139mol的化合物a(4g)加到250ml三口瓶中,再加入100ml甲胺的四氢呋喃溶液,搅拌,氮气保护,冰盐浴降温至内温为-5℃,开始缓慢滴加0.0139mol四氯化钛(2.6g),控制反应温度不高于5℃,并在20分钟滴完;所述甲胺的四氢呋喃溶液中,甲胺的物质的量浓度为2mol/l;(1-2)滴完后,升温至20℃-25℃反应4小时,再升温至40℃-45℃反应1小时;反应过程中,采用tlc监控反应,原料消失,停止反应;(1-3)反应结束后,向反应液中滴加50ml水猝灭反应,再加入50ml水和50ml乙酸乙酯,萃取分液,保留上层有机相;(1-4)上层有机相用饱和食盐水洗涤一次,然后用无水硫酸钠干燥;(1-5)过滤,并用乙酸乙酯冲洗滤饼两次,乙酸乙酯每次用量为20ml,合并滤液和洗液,减压浓缩干,得到4.1g化合物b,化合物b为淡黄色固体。(2)由化合物b合成化合物c;(2-1)0.0136mol化合物b(4.1g)加到150ml三口瓶中,依次加入25ml乙酸和5ml水,搅拌,冰水浴降温至内温3℃,开始缓慢滴加7.2ml亚硝酸钠水溶液,亚硝酸钠的物质的量浓度为4.72mol/l,15分钟滴完;(2-2)滴完后,升温至25℃反应3小时;(2-3)反应结束后,向反应液中加入40ml乙酸乙酯,再滴加饱和碳酸钠水溶液调节ph值至8;(2-4)然后分液,保留有机相,水相再用40ml乙酸乙酯萃取一次,合并有机相,然后用50ml饱和食盐水洗涤有机相,用无水硫酸钠干燥;(2-5)过滤,并用乙酸乙酯冲洗滤饼2次,乙酸乙酯每次为15ml,合并滤液和洗液,减压浓缩干,层析柱纯化,淋洗剂为石油醚pe和乙酸乙酯ea的混合物,pe与ea的体积比为20:1,收集产品,得到1.5g化合物c,化合物c为黄色固体。(3)由化合物c合成氟氯唑仑化合物d:(3-1)4.544mmol化合物c(1.5g)加到100ml单口瓶中,依次加入20mln,n-二甲基甲酰胺dmf、2.499mmol对甲苯磺酸(430mg)和5.453mmol乙酰肼(404mg),加热至120℃反应4小时,tlc监控反应;(3-2)停止反应,降温至室温,加入40ml饱和碳酸氢钠水溶液和30ml二氯甲烷,萃取分液,保留有机相,水相再用二氯甲烷萃取2次,二氯甲烷每次用量为20ml,合并有机相;然后用50ml饱和食盐水洗涤有机相,用无水硫酸钠干燥;(3-3)过滤,并用二氯甲烷冲洗滤饼2次,二氯甲烷每次用量为10ml,合并滤液和洗液,减压浓缩干,层析柱纯化,淋洗剂为二氯甲烷和甲醇的混合液,二氯甲烷和甲醇的体积比为50:1,收集纯度高的产品点,得到372mg黄色液体的化合物d粗品;(3-4)化合物d粗品溶于3ml二氯甲烷,再缓慢滴加15ml异丙醚,有固体析出,过滤,并用异丙醚冲洗滤饼2次,异丙醚每次为4ml,取滤饼,抽干,得到280mg化合物d,化合物d为白色固体。三、结果与讨论1.核磁共振谱图表征1.1化合物d氟氯唑仑的核磁共振氢谱如图3所示:1hnmr(400.13mhz,cdcl3,297k):7.69,7.67(dd,jh-h=7.7hz,1h,c10h),7.65,7.62(dd,jh-h=8.8hz,1h,c14h),7.48-7.46(m,1h,c15h),7.43(d,jh-h=8.6hz,c11h),7.35(d,1h,j=2.6hz,c8h),7.28-7.24(m,1h,c17h),7.06-7.01(m,1h,c16h),5.65(d,jh-h=12.8hz,1h,h1),4.14(d,jh-h=12.8hz,1h,h2),2.65(s,1h,ch3).1.2化合物d氟氯唑仑的核磁共振碳谱如图4所示。13c{1h}nmr(100.6mhz,cdcl3,297k):165.02(c9),161.51(c13),159.01(c12),154.77(c4),150.43(c18),133.67(c2),132.71(d,c15),131.79(d,c14),131.10(c10),130.92(c1),130.05(c8),127.14(d,c7),124.67(c17),124.52(c11),116.40(d,c16),46.40(c5),12.28(c23)ppm.1.3化合物d氟氯唑仑的1h/13chmbc、1h/13chmqc和1h/1hcoesy谱图由图3和图4,以及图5的hmbc、图6的hmqc和图7coesy所示的二维nmr图谱进行进一步解析,归属了所有的碳和氢信号,见图2。1.4化合物d氟氯唑仑的核磁共振氟谱如图8所示:19f{1h}nmr(376.5mhz,cdcl3,297k):-112.1ppm.2.化合物d氟氯唑仑液质联用谱图化合物d氟氯唑仑的lc-q-tof-ms的tic及eic谱图,如图9a和图9b所示。配置浓度为10ng/ml氟氯唑仑,溶剂为乙腈,进样量为1.000μl。具体条件为:色谱条件:agilenteclipsec18色谱柱(2.1mm×150mm,1.8μm),柱温:35℃;流动相为a:水+10mmol/l乙酸铵+0.1vt%甲酸,b:乙腈+0.1vt%甲酸。梯度洗脱程序见表1。表1time(min)flow(ml/min)a%b%00.3901010.390105.50.301008.50.301008.60.39010180.39010质谱条件:电喷雾电离-正离子模式(esi+),vcap:+3000v,气体温度:350℃,雾化气:n2,35psi,干燥气:n2,10l/min。flagmentor:125v,skimmer:60v;ms/ms碰撞气:he,碰撞能:33v。如图10a和10b所示为lc-q-tof-ms在正离子模式下的一级质谱图和二级质谱图。3.化合物d氟氯唑仑高效液相色谱图配置浓度为0.5mg/ml氟氯唑仑,溶剂为乙腈,进样量为1.000μl。具体条件为:色谱条件:agelatechnologiesvenusilmpc18色谱柱(4.6mm×150mm,5.0μm),柱温:30℃;流动相为a:水+0.02vt%甲酸,b:乙腈+0.02vt%甲酸,检测波长:254nm。梯度洗脱程序见表2。表2time(min)flow(ml/min)a%b%01.095514.51.059514.91.0955161.0955氟氯唑仑的hplc谱图如图11所示,测试结果如表3所示。表3由图11和表3可知,氟氯唑仑的纯度为99.1%。可直接用作公安机关检验鉴定工作的对照品。显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本专利申请权利要求的保护范围之中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1