一种含有索拉非尼的药物组合物及其应用的制作方法
本发明属于生物医药技术领域,具体涉及通过联合用药治疗恶性肿瘤,特别涉及索拉非尼和一种新的化合物的协同作用而治疗恶性肿瘤。
背景技术:
恶性肿瘤是导致人类疾病和死亡的重要顽疾之一,它严重危害人类的健康和生命安全。在我国,常发性恶性肿瘤以肺癌、胃癌、肝癌、乳腺癌为主。癌症的传统治疗有手术、放疗、化疗。近年来,随着细胞和分子生物学研究的不断深入,研发用于治疗恶性肿瘤的药物,尤其是筛选高效低毒,专门针对肿瘤细胞特定信号通路或靶位点的药物越来越受到关注,并被认为是最终根治恶性肿瘤的重要方向。
正常细胞的癌变总是伴随着核酸和蛋白质合成的显著增加。而核酸和蛋白质的高速合成也是肿瘤细胞快速增长的必要前提。这些肿瘤细胞的大分子合成过程都需要不断提供必需和非必需的氨基酸。作为人体中最丰富的氨基酸的谷氨酰胺,在肿瘤细胞的生长和肿瘤生成过程中起到了至关重要的作用[参见文献:1-3]。而谷氨酰胺酶则是谷氨酰胺新陈代谢的过程中的关键一环。谷氨酰胺酶位于细胞中线粒体的内膜[参见文献:4],可以催化由谷氨酰胺生成谷氨酸的反应,谷氨酸在谷氨酸脱氢酶的作用下转变为α-酮戊二酸,以底物的形式进入三羧酸循环,为肿瘤细胞的大分子合成提供新陈代谢的中间体[参见文献:5]。科学研究证明谷氨酰胺酶的高活性和肿瘤细胞的快速生长紧密相关[参见文献:6]。用谷氨酰胺酶的反义(antisense)mrna去转染艾氏腹水癌细胞,不但它们的生长受到抑制而且形态也发生了变化。用反义mrna转染的癌细胞,接种到小鼠体内,这样的癌细胞完全失去了产生肿瘤的能力,而这样的小鼠也和健康动物完全一样[参见文献:7]。这些科学发现充分说明了谷氨酰胺酶的活性与肿瘤发生和发展紧密相关。因此,谷氨酰胺酶已成为抗肿瘤疗法中受到人们极大关注的目标蛋白。近年来,一些小分子谷氨酰胺酶抑制剂如二噻二唑类化合物bptes(bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethylsulfide)[参见文献:8],杂环类化合物cb-839(2-(pyridin-2-yl)-n-(5-(4-(6-(2-(3-(trifluoromethoxy)phenyl)acetamido)pyridazin-3-yl)butyl)-1,3,4-thiadiazol-2-yl)acetamide)被筛选了出来,并作为潜在的抗肿瘤药物在美国进入了临床实验阶段。但是,迄今为止,在动物实验或临床测试中,这些谷氨酰胺酶小分子抑制剂在单独使用时对肿瘤生长的抑制效果却非常有限。
正常细胞的癌变之外,在恶性肿瘤生成过程中,细胞癌变后会在其周围诱导血管形成(即tumorangiogenesis,肿瘤血管生成),这是肿瘤生长中的关键一环。一方面,肿瘤需要通过这些新生血管为自身提供快速生长所需要的养分,另一方面,肿瘤细胞也能通过这些血管进入循环从而转移扩散。因此,抗肿瘤治疗的关键策略之一就是抑制肿瘤血管生成。现今,抑制肿瘤血管生成的药物有:贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche);索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);珀玛利度胺(pomalidomide,商品名:pomalyst,celgene)等等。抑制肿瘤血管生成常常是通过使用靶向药物抑制肿瘤血管发育过程中的关键因子,例如血管内皮生长因子(vascularendothelialgrowthfactor,vegf)来达成。抗vegf抗体,例如贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche)就是第一个被fda批准用于肿瘤临床治疗的抑制肿瘤血管生成的药物。然而,贝伐单抗(以及其它靶向vegf信号的药物)有明显的副作用,它们本身也并不直接抑制肿瘤细胞的生长。而且临床上大多数病人会产生耐药性[参见文献:9-13]。因此,肿瘤血管生成抑制剂这类药物,如贝伐单抗,索拉非尼,舒尼替尼等,对相当部分病人来说是费用高昂而效果并不显著的。
技术实现要素:
本发明通过提供一种一定有效量的谷氨酰胺酶抑制剂与肿瘤血管生成抑制剂联合用药,能有效抑制肿瘤细胞的快速生长,肿瘤周围的血管生成和肿瘤的转移,对肿瘤治疗具有显著疗效,所取得的抗肿瘤作用明显优于单一用药,具有显著的协同和增效作用,是一类高效低毒的抗肿瘤药物组合。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂如通式(i)所示或其生理上可以接受的盐:
其中:
r0,r0’独立地选自哒嗪基:
或者以下通式:
x独立地选自s、o、c;优选为s、c;更优选为s;ch单元的任何氢原子可被烷基取代。
y独立地选自n、o、s,c;优选为n,o;更优选为n;nh或ch单元的任何氢原子可被烷基取代。
r1,r2均独立地选自h或r3(co):
r3独立地选自h,oh,芳基,杂芳基,杂芳基烷基,杂芳氧基,(c1-c6)烷基,o-(c1-c6)烷基,o-(c1-c6)烷基-cooh,o-(c1-c6)烷基-芳基,oco-(c1-c6)烷基,s-(c1-c6)烷基-芳基,no2>nh2>nh-(c1-c6)烷基,n((c1-c6)烷基)2,n-(c1-c6)烷基-芳基;
其中任何游离羟基可以被酰化,以形成-c(o)r4;优选为芳基,杂芳基,s-(c1-c6)烷基-芳基,n-(c1-c6)烷基-芳基,o-(c1-c6)烷基-芳基;更优选为芳基(苯基),例如乙苯;r4独立地选自h或取代烷基,烷氧基,氨基烷基,烷基氨基烷基,杂环基烷基,芳烷基,或杂环的烷氧基。
本文所使用的术语“芳基”指其中至少一个环为芳香族的c6-12单环烃环或双环烃环。所述基团的实例包括苯基、萘基和四氢萘基。
本文所使用的术语“杂芳基”是指5-6元芳香单环或稠合的8-10元芳香双环,所述单环或双环包含选自氧、氮和硫的1至4个杂原子。这类芳香单环的实例包括噻吩基、呋喃基、呋吖基(furazanyi)、吡咯基、三唑基、四唑基、咪唑基、噁唑基、噻唑基、噁二唑基、异噻唑基、异噁唑基、噻二唑基、吡喃基、吡唑基、嘧啶基、哒嗪基、吡嗪基、吡啶基、三嗪基、四嗪基等。这类芳香双环的实例包括喹啉基、异喹啉基、喹唑啉基、喹喔啉基、蝶啶基、噌啉基、酞嗪基(phthalazinyl)、萘唳基(naphthyridinyl)、引哚基、异氮却基、氮杂引哚基(azaindolyl)、吲嗪基(indolizinyl)、吲唑基、嘌呤基、吡咯并吡啶基、呋喃并吡啶基、苯并呋喃基、异苯并呋喃基、苯并噻吩基、苯并咪唑基、苯并噁唑基、苯并异噁唑基、苯并噻唑基、苯并异噻唑基、苯并噁二唑基、苯并噻二唑基和咪唑并吡啶基。
本文所使用的术语“(c1-c6)烷基”是指含1至6个碳原子的直链饱和烃基团或支链饱和经基团。(c1-c6)烷基基团的实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、戊基、己基。优选地,所述烃基团为直链的。
当本发明化合物是以不同的对映异构体和/或非对映异构体形式存在时(包括双键的几何异构),所述化合物可制备为同分异构混合物或外消旋化合物,但本发明涉及所有这类对映异构体或同分异构体,不论是以光学纯形式或作为与其他同分异构体的混合物存在。独立的对映异构体或同分异构体可通过本领域已知的方法获得,例如产物或中间体的光学分辨(例如手性色谱分离(例如,手性hplc)),或对映异构体合成方法。类似地,当本发明化合物可作为替代的互变异构体形式存在(例如,酮/烯醇、酰胺/亚氨酸)时,本发明涉及分离的独立互变异构体,并且涉及所有比例的互变异构体混合物。
本发明还涉及上述通式化合物的可药用盐,所述可药用盐是本领域技术人员熟知的,包括无机酸和有机酸的碱式盐,或者无机碱或有机碱的酸式盐。所述酸例如盐酸、氢溴酸、硫酸、磷酸、甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸、乙酸、三氟乙酸、苹果酸、酒石酸、柠檬酸、乳酸、草酸、富马酸、琥珀酸、马来酸、苯甲酸等。所述碱例如有碱金属或碱土金属阳离子,或铵阳离子等。
所述的本发明的化合物可通过一般性制备方法或者其他已知方法[参见文献:14]获得。在制备所述化合物中使用的起始物质和试剂可从供应商处购得或者可通过本领域技术人员已知的方法制备。所述一般性路线仅仅举例说明了通过其可合成本发明化合物的方法,并且对于已经参考本公开内容的本领域技术人员,所述路线的多种修改是可以做出的并且是得到启示的。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是bptes(bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethylsulfide)。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gb-002,如式(ii)所示:
2-苯基-n-(6-((1-(6-(2-(3-(三氟甲氧基)苯基)乙酰氨基)哒嗪-3-基)哌啶-4-基)氨基)哒嗪-3-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gb-016,如式(iii)所示:
2-苯基-n-(5-((1-(6-(2-(3-(三氟甲氧基)苯基)乙酰氨基)哒嗪-3-基)哌啶-4-基)氨基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gb-017,如式(iv)所示:
2-苯基-n-(5-((1-(6-(2-(3-(三氟甲氧基)苯基)乙酰氨基)哒嗪-3-基)哌啶-4-基)氧基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gb-042,如式(v)所示:
2-苯基-n-(5-(1-(5-(2-(3-(三氟甲氧基)苯基)乙酰氨基)-1,3,4-噻二唑-2-基)哌啶-4-基氨基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gb-051,如式(vi)所示:
2-(吡啶-4-基)-n-(5-(1-(5-(2-(4-(三氟甲氧基)苯基)乙酰氨基)-1,3,4-噻二唑-2-基)哌啶-4-基氨基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-002,如式(vii)所示:
n-(5-(1-(5-(2-(1h-吡唑-1-基)乙酰氨基)-1,3,4-噻二唑-2-基)哌啶-4-基氨基)-1,3,4-噻二唑-2-基)-2-(1h-吡唑-1-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-003,如式(viii)所示:
2-苯基-n-(5-(4-((5-(2-苯基乙酰氨基)-1,3,4-噻二唑-2-基)氨基)哌啶-1-基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-005,如式(ix)所示:
2-苯基-n-(5-(4-((5-(2-苯基乙酰氨基)-1,3,4-噻二唑-2-基)氧基)哌啶-1-基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-006,如式(x)所示:
n2-苯乙基-n5-(1-(5-(苯乙胺)-1,3,4-噻二唑-2-基)哌啶-4-基)-1,3,4-噻二唑-2,5-二胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-024,如式(xi)所示:
2-(4-氨基苯基)-n-(5-(4-(5-(2-苯基乙酰氨基)-1,3,4-噻二唑-2-基氨基)哌啶-1-基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-036,如式(xii)所示:
2-(2-氯嘧啶-5-基)-n-(5-(1-(5-(2-(4-(三氟甲氧基)苯基)乙酰氨基)-1,3,4-噻二唑-2-基)哌啶-4-基氨基)-1,3,4-噻二唑-2-基)乙酰胺。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述肿瘤血管生成抑制剂是贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche);或索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);或舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);或珀玛利度胺(pomalidomide,商品名:pomalyst,celgene)。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是bptes(bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethylsulfide),所述肿瘤血管生成抑制剂是贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche);或索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);或舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);或珀玛利度胺(pomalidomide,商品名:pomalyst,celgene)。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gb-016,所述肿瘤血管生成抑制剂是珀玛利度胺(pomalidomide,商品名:pomalyst,celgene);或索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);或舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);或贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche)。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-003,所述肿瘤血管生成抑制剂是贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche);或索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);或舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);或珀玛利度胺(pomalidomide,商品名:pomalyst,celgene)。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-005,所述肿瘤血管生成抑制剂是珀玛利度胺(pomalidomide,商品名:pomalyst,celgene);或索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);或舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);或贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche)。
本发明所提供的抗恶性肿瘤的药物组合物,其特征在于,所述谷氨酰胺酶抑制剂是gi-006,所述肿瘤血管生成抑制剂是索拉非尼(sorafenib,商品名:蕾莎瓦,nexavar,bayer);或舒尼替尼(sunitinib,商品名:索坦,sutent,pfizer);或珀玛利度胺(pomalidomide,商品名:pomalyst,celgene);或贝伐单抗(bevacizumab,商品名:阿瓦斯丁,avastin,genentech/roche)。
本发明的另一个目的是提供所述的药物组合物在制备治疗肿瘤疾病的药物中的应用。
本发明所提供的抗恶性肿瘤的药物制剂,是以上述抗恶性肿瘤的药物组合物作为活性成分,并辅以可接受的药用辅料、载体和/或赋形剂,以制成各种制剂,如,但不限于,口服液、胶丸、口服混悬液、固体分散体、胶囊、缓释剂、颗粒剂、片剂、注射剂、软膏、贴剂、植入剂等。
可用于本发明的药用组合物中的可药用载体、助剂和介质是在药用制剂领域中常规使用的那些,包括但不限于,糖、糖醇、淀粉、离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白(例如人血清白蛋白)、缓冲物质(例如磷酸盐)、甘油、山梨酸、山梨酸钾、饱和植物脂肪酸的部分甘油酯混合物、水、盐或电解质(例如硫酸鱼精蛋白)、磷酸氢二钠、磷酸氢钾、氯化钠、锌盐、胶态氧化硅、三硅酸镁、聚乙烯比咯烷酮、基于纤维素的物质、聚乙二醇、羧甲基纤维素钠、聚丙烯酸酯、蜡、聚乙烯-聚环氧丙烷-嵌段聚合物、聚乙二醇和羊毛脂。
本发明的组合制剂的组成成分可以同时、分开或顺序给药。
本发明的组合制剂可用于针对在包括人类的哺乳动物中发现的包括肺、白血病、黑色素瘤、肝脏、乳腺、卵巢、前列腺、胃、胰腺、肾脏、结肠和中枢神经系统肿瘤或癌症的所有类型的治疗中。优选地,用于非小细胞肺癌、乳腺癌、神经胶质瘤、肝癌的治疗。
本发明的药物组合物还可能包含一种或多种另外的活性药物组分。本发明的药物组合可与一种或多种另外的活性药物组分一块给药。该组合物可以是含本发明药物组合物和一种或多种另外的活性药物组分的单一组合物的形式。或者,该组合物可为两种或多种单独的组合物组合的形式,其中本发明的药物组合包含在一种组合物中,一种或多种另外的活性药物组分包含在一种或多种单独的组合物中。在所述的药物组合物中,所述另外的活性药物组分例如可以是另一种抗肿瘤药物。
本发明所提供的化学结构式如式i所示的谷氨酰胺酶抑制剂,在制备治疗或预防与谷氨酰胺酶活性增高有关病症的药物中的用途。其中,谷氨酰胺酶抑制剂是gb-002、gb-016、gb-017、gb-042、gb-051、gi-002、gi-003、gi-005、gi-006、gi-024、gi-036。
本发明所用术语“一定有效量的”,是指抗恶性肿瘤的药物组合物中的各组分的量,该量足以实现所需的抑制肿瘤细胞的快速生长,肿瘤周围的血管生成和肿瘤的转移的效果的,均属于该发明范畴。本发明研究也表明不同的肿瘤细胞类型对该组合物的敏感程度是有区别的。
本发明在具体的动物实验中,应用多个将来自于病人的肿瘤组织植入严重联合免疫缺陷小鼠(severecombinedimmunodeficiencymice,scidmice)建立的人源肿瘤株系小鼠模型(patient-derivedxenograftmicemodel,pdxmodel),谷氨酰胺酶抑制剂和肿瘤血管生成抑制剂的联合用药在剂量仅为两种抑制剂单一使用时剂量的三分之一到五分之一的情况下,对这些肿瘤株生长的抑制高达90%以上。而两种抑制剂单一使用时对这些肿瘤株生长的抑制仅约5%-20%。免疫组织化学的实验也表明,上述的联合用药显著抑制了这些人源肿瘤周围血管的生成和肿瘤向肺部转移。与此相对照,同样条件下,两种抑制剂单一使用时对肿瘤周围血管的生成和肿瘤向肺部转移均无显著影响。在类似的动物实验中,对肝癌肿瘤株,神经胶质瘤肿瘤株,乳腺癌肿瘤株的研究均得到了相似的结果。
基于以上的系列研究结果,并考虑到联合用药的剂量仅为单一使用时剂量的五分之一,相应副作用远低于单一用药。因此,一定比例的谷氨酰胺酶抑制剂和肿瘤血管生成抑制剂联合用药对肿瘤治疗具有显著疗效,相比于单一用药有显著的协同和增效作用,是一类高效低毒的抗肿瘤药物组合。本发明相关的分子生物学研究也阐明了这一类药物组合具有显著的协同增效作用的分子机理。
附图说明
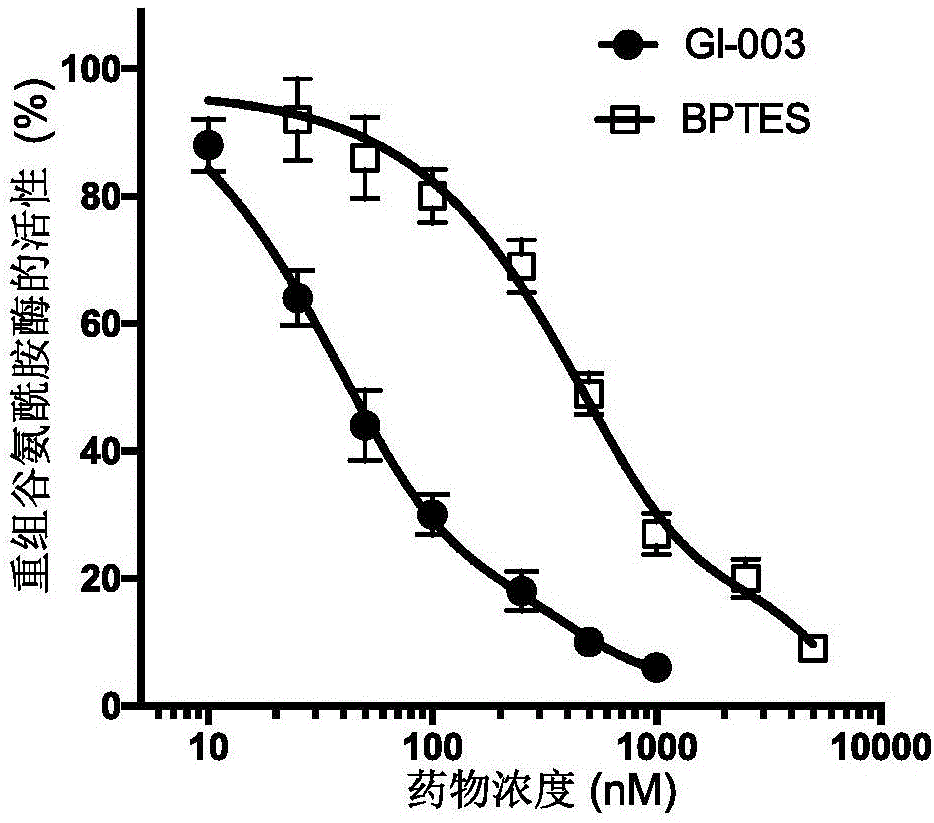
图1是bptes和gi-003抑制重组谷氨酰胺酶活性;
图2是bptes和gi-003抑制肿瘤细胞mda-mb231中谷氨酰胺酶活性;
图3是gi-003抑制肿瘤细胞的生长;
图3a显示出gi-003对非小型肺癌细胞crl-5803(atcc)的生长抑制;
图3b显示出gi-003对乳腺癌细胞mda-mb231的生长抑制;
图3c显示出gi-003对人神经胶质瘤细胞u87(atcc)的生长抑制;
图4是gi-003对正常乳腺上皮细胞hmec生长的影响;
图5是谷氨酰胺酶抑制剂和肿瘤血管生成抑制剂药物组合对小鼠体内肿瘤的抑制作用;
图5a显示出贝伐单抗和bptes药物组合对小鼠体内乳腺癌细胞株mda-mb231生成肿瘤的生长的抑制作用;
图5b显示出珀玛利度胺和gi-005药物组合对小鼠体内乳腺癌细胞株mda-mb231生成肿瘤的生长的抑制作用;
图5c显示出索拉非尼和gi-006药物组合对小鼠体内乳腺癌细胞株mda-mb231生成肿瘤的生长的抑制作用;
图5d显示出gi-003和贝伐单抗药物组合对小鼠体内人肺癌细胞株a549生成肿瘤的生长的抑制作用;
图5e显示出gi-003和贝伐单抗药物组合对小鼠体内人肺癌细胞株a549生成肿瘤的肿瘤血管生成的抑制作用;
图6是谷氨酰胺酶抑制剂和肿瘤血管生成抑制剂药物组合对小鼠体内植入的人源肿瘤组织建立的肿瘤株系(pdxmodel)的抑制作用;
图6a显示出gi-003和贝伐单抗药物组合对小鼠体内植入的人源肺癌肿瘤株系hlc-3(humanlungcancer-3)生成肿瘤的生长的抑制作用;
图6b显示出bptes和贝伐单抗药物组合对小鼠体内植入的乳腺癌肿瘤株系hbc-2(humanbreastcancer-2)生成肿瘤的生长的抑制作用;
图6c显示出gi-003和贝伐单抗药物组合对小鼠体内植入的人源肺癌肿瘤株系hlc-3的肿瘤血管生成的抑制作用;
图6d显示出bptes和贝伐单抗药物组合对小鼠体内植入的乳腺癌肿瘤株系hbc-2的肿瘤血管生成的抑制作用。
具体实施方式
通过以下详细说明结合附图可以进一步理解本发明的特点和优点。所提供的实施例仅是对本发明方法的说明,而不以任何方式限制本发明揭示的其余内容。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。
除非另有说明,本说明书中使用的全部专业术语和科学用语的含义均与本发明所属技术领域的技术人员一般理解的含义相同。但如有冲突,以包含定义的本说明书为准。
【实施例1】谷氨酰胺酶抑制剂化合物gi-003(glutaminaseinhibitor-003)的制备
本实施例提供了谷氨酰胺酶抑制剂gi-003的制备方法,具体步骤为:
(1)将等克分子的氨基硫脲和乙氧基乙酸在三氯氧化磷的催化下,90℃迴流3小时。将反应混合物冷却至室温,加冰水,用氢氧化钠调ph=14。白色沉淀用水洗涤。真空干燥。即为中间产物【1】
5-(ethoxymethyl)-1,3,4-thiadiazol-2-amine
(5-(乙氧基甲基)-1,3,4-噻二唑-2-胺)
(2)将中间产物【1】与等克分子的苯乙酰氯和吡啶混合,加热至混合物均匀,溶液冷却至室温,加入甲醇后过滤,留下粗的固体,即为中间产物【2】
n-(5-(hydroxymethyl)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide
(n-(5-(乙氧基甲基)-1,3,4-噻二唑-2-基)-2-苯乙酰胺)
(3)将中间产物【2】在硫酸介质下,用溴化氢(或碘化氢)使醚链裂解,生成醇,再加入重铬酸钾和硫酸,使之进一步氧化的生成酸,再加入乙酸(80℃-90℃)迴流氧化脱去杂环上的c,用甲醇沉淀过滤,即为中间产物【3】
n-(5-hydroxy-1,3,4-thiadiazol-2-yl)-2-phenylacetamide
(n-(5-羟基-1,3,4-噻二唑-2-基)-2-苯乙酰胺)
(4)将中间产物【3】加入相当于中间产物【3】的克分子量一半的对氨基哌啶。在硫酸介质下再加热迴流1-2小时。反应产物加甲醇沉淀过滤。粗固体用二甲亚砜重结晶,即为最终产物gi-003(viii)。
2-phenyl-n-(5-(4-((5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)amino)piperidin-1-yl)-1,3,4-thiadiazol-2-yl)acetamide
2-苯基-n-(5-(4-((5-(2-苯基乙酰氨基)-1,3,4-噻二唑-2-基)氨基)哌啶-1-基)-1,3,4-噻二唑-2-基)乙酰胺
【实施例2】:谷氨酰胺酶抑制剂化合物gi-006(glutaminaseinhibitor-006)的制备
本实施例提供了谷氨酰胺酶抑制剂gi-006的制备方法,具体步骤为:(1)将等克分子的氨基硫脲和乙氧基乙酸在三氯氧化磷的催化下,90℃迴流3小时。将反应混合物冷却至室温,加冰水,用氢氧化钠调ph=14。白色沉淀用水洗涤。真空干燥。即为中间产物【1】
5-(ethoxymethyl)-1,3,4-thiadiazol-2-amine
(5-(乙氧基甲基)-1,3,4-噻二唑-2-胺)
(2)将中间产物【1】与等克分子的苯乙酰氯和吡啶混合,加热至混合物均匀,溶液冷却至室温,加入甲醇后过滤,留下粗的固体,即为中间产物【2】
n-(5-(hydroxymethyl)-1,3,4-thiadiazol-2-yl)-2-phenylacetamide
(n-(5-(乙氧基甲基)-1,3,4-噻二唑-2-基)-2-苯乙酰胺)
(3)将中间产物【2】在硫酸介质下,用溴化氢(或碘化氢)使醚链裂解,生成醇,再加入重铬酸钾和硫酸,使之进一步氧化的生成酸,再加入乙酸(80℃-90℃)迴流氧化脱去杂环上的c,用甲醇沉淀过滤,即为中间产物【3】
n-(5-hydroxy-1,3,4-thiadiazol-2-yl)-2-phenylacetamide
(n-(5-羟基-1,3,4-噻二唑-2-基)-2-苯乙酰胺)
(4)将中间产物【3】与适量的氢氧化钠溶液,肼和二乙二醇醚一起加热,使肼变成腙。然后将水和多余的肼蒸出,待温度达到腙开始分解的温度(195℃-200℃)时,再迴流,使n2放出。在常压下,反应均为3-5小时。产物可用甲醇沉淀过滤,即得中间产物【4】
5-(phenethylamino)-1,3,4-thiadiazol-2-ol
(5-(苯乙基氨基)-1,3,4-噻二唑-2-醇)
(5)将中间产物【4】加入相当于中间产物【4】的克分子量一半的对氨基哌啶。在硫酸介质下再加热迴流1-2小时。反应产物加甲醇沉淀过滤。粗固体用二甲
亚砜重结晶,即为最终产物gi-006(x)。
n2-phenethyl-n5-(1-(5-(phenethylamino)-1,3,4-thiadiazol-2-yl)piperidin-4-yl)-1,3,4-thiadiazole-2,5-diamine
(n2-苯乙基-n5-(1-(5-(苯乙胺)-1,3,4-噻二唑-2-基)哌啶-4-基)-1,3,4-噻二唑-2,5-二胺)
【实施例3】化合物gi-003对重组谷氨酰胺酶活性的抑制
在大肠杆菌中表达并制备小鼠谷氨酰胺酶(分子量为66kd)的重组蛋白,用不同浓度的化合物gi-003或者bptes处理后测其酶活性。具体步骤如下:使用克隆有n-端连有组氨酸的小鼠谷氨酰胺酶的基因的pet28a质粒(novagen:目录号:69864-3)在大肠杆菌中表达小鼠谷氨酰胺酶。该重组谷氨酰胺酶蛋白用阴离子交换柱层析做进一步纯化。将1μm谷氨酰胺酶重组蛋白在57μmtris-乙酸(ph8.6)和0.25μmedta的缓冲液中与不同浓度的化合物gi-003或者用作对照的已知的谷氨酰胺酶抑制剂bptes—起温浴,终体积为80μl,旋转30分钟。化合物gi-003或者bptes用dmso稀释,使加入的体积在不同的反应中保持恒定(5μl)。然后加入谷氨酰胺使终体积为115μl,终浓度为17μm,反应在37℃进行1小时,然后加入10μl3m氯化氢终止反应。从第一个反应中取出10μl加入到第二个反应中,第二个反应包含了114μmtris-hcl(ph9.4),0.35μmadp,1.7μmnad和6.3u/ml的谷氨酸脱氢酶,终体积为228μl。第二个反应在室温下进行45分钟,然后在340nm的波长下测定其吸收值,以计算谷氨酰胺酶的活性。结果显示,化合物gi-003对谷氨酰胺酶活性具有显著的抑制作用,并且其抑制作用远优于己知的谷氨酰胺酶抑制剂bptes:其ic50约为50nm,远低于bptes(图1)。
【实施例4】化合物gi-003对肿瘤细胞中谷氨酰胺酶活性的抑制
从相同数量的乳腺癌细胞mda-mb231或神经胶质瘤细胞u87中分离线粒体,肿瘤细胞经不同浓度的化合物gi-003或者bptes处理,对不同情况下的细胞中分离的线粒体做谷氨酰胺酶的活性的测定。图2显示的是mda-mb231细胞中谷氨酰胺酶活性的测定结果。结果显示,化合物gi-003对谷氨酰胺酶活性具有显著的抑制作用,并且其抑制作用远优于己知的谷氨酰胺酶抑制剂bptes(图2)。
【实施例5】化合物gi-003对肿瘤细胞生长的抑制作用
将非小型肺癌细胞crl-5803,乳腺癌细胞mda-mb231或神经胶质瘤细胞u87培养在rpmi1640的培养液中,加入10%的胎牛血清,同时用化合物gi-003(浓度为250nm)或者dmso处理细胞,在1,2,4,6天时进行细胞计数(细胞生长定量)。结果如图3a,3b,及3c所示,本发明所述化合物gi-003均可抑制crl-5803,mda-mb231,或u87细胞的生长。
【实施例6】化合物gi-003对正常细胞的影响
将人类正常乳腺上皮细胞hmec培养在megm(lonza)完全培养液中,用250nμ化合物gi-003或者dmso处理,分别在1,2,4,6,8天进行细胞计数(细胞生长定量)。结果显示,化合物gi-003对于正常hmecs的生长没有明显的影响(图4)。
【实施例7】本发明涉及的药物组合物对小鼠体内肿瘤的抑制作用
7.1、bptes/贝伐单抗对小鼠体内乳腺癌细胞株生成肿瘤的抑制作用
将乳腺癌细胞mda-mb231(3×106)皮下注射到严重联合免疫缺陷小鼠(scidmice,nationalcancerinstitute)的胁腹,18天后肿瘤长到约200mm3,开始用谷氨酰胺酶抑制剂bptes或肿瘤血管生成抑制剂贝伐单抗,或者二者联合使用处理肿瘤,其方法是:每隔一天腹腔注射200μl的谷氨酰胺酶抑制剂bptes(2mg/kg小鼠体重),贝伐单抗(2mg/kg小鼠体重),或者二者联合,持续处理10天。对照动物用同样的方法注射不含药物的同样助溶载体。在此期间,毎2-3天测定肿瘤大小,计算肿瘤的体积[长(mm)×宽(mm)×高(mm)/2]。结果显示(图5a),10天后,与对照组相比,谷氨酰胺酶抑制剂或贝伐单抗单独使用时,对肿瘤生长的抑制分别约为5%和16%,而二者联用时对肿瘤生长的抑制高达90%。说明该药物组合物腹腔注射入小鼠时有显著加成效果,可到达肿瘤而对肿瘤起到积极的治疗作用。
7.2、gi-005/珀玛利度胺对小鼠体内乳腺癌细胞株生成肿瘤的抑制作用
将乳腺癌细胞mda-mb231(3×106)皮下注射到严重联合免疫缺陷小鼠(scidmice)的胁腹,18天后肿瘤长到约200mm3,开始用谷氨酰胺酶抑制剂gi-005或肿瘤血管生成抑制剂珀玛利度胺(pomalidomide),或者二者联合使用处理肿瘤,其方法是:每隔一天腹腔注射200μlgi-005(2mg/kg小鼠体重),珀玛利度胺(0.5mg/kg小鼠体重),或者二者联合,持续处理10天。对照动物用同样的方法注射不含药物的同样助溶载体。在此期间,毎2-3天测定肿瘤大小,计算肿瘤的体积。结果显示(图5b),10天后,与对照组相比,谷氨酰胺酶抑制剂或珀玛利度胺单独使用时,对肿瘤生长的抑制分别约为2%和40%,而二者联用时对肿瘤生长的抑制达80%。
7.3、gi-006/索拉非尼对小鼠体内乳腺癌细胞株生成肿瘤的抑制作用
将乳腺癌细胞mda-mb231(3×106)皮下注射到严重联合免疫缺陷小鼠(scidmice)的胁腹,18天后肿瘤长到约200mm3,开始用谷氨酰胺酶抑制剂gi-006或肿瘤血管生成抑制剂索拉非尼(sorafenib),或者二者联合使用处理肿瘤,其方法是:每隔一天腹腔注射200μl的谷氨酰胺酶抑制剂gi-006(2mg/kg小鼠体重),索拉非尼(20mg/kg小鼠体重),或者二者联合,持续处理10天。对照动物用同样的方法注射不含药物的同样助溶载体。在此期间,毎2-3天测定肿瘤大小,计算肿瘤的体积。结果显示(图5c),10天后,与对照组相比,谷氨酰胺酶抑制剂或索拉非尼单独使用时,对肿瘤生长的抑制分别约为7%和25%,而二者联用时对肿瘤生长的抑制达70%。
7.4、gi-003/贝伐单抗对小鼠体内肺癌细胞株生成肿瘤的抑制作用
将肺癌细胞a549(3×106)皮下注射到严重联合免疫缺陷小鼠(scidmice)的胁腹,24天后肿瘤长到约200mm3,开始用谷氨酰胺酶抑制剂gi-003或肿瘤血管生成抑制剂贝伐单抗,或者二者联合使用处理肿瘤,其方法是:每隔一天腹腔注射200μl的gi-003(0.5mg/kg小鼠体重),贝伐单抗(2mg/kg小鼠体重),或者二者联合,持续处理12天。对照动物用同样的方法注射不含药物的同样助溶载体。在此期间,毎2-3天测定肿瘤大小,计算肿瘤的体积[长(mm)×宽(mm)×高(mm)/2]。结果显示(图5d),与对照组相比,谷氨酰胺酶抑制剂gi-003或贝伐单抗单独使用时,对肿瘤生长的抑制分别约为5%和15%,而二者联用时对肿瘤生长的抑制高达85%。将实验中生成的肿瘤及周边组织取出,切片后进行免疫组化研究。使用抗cd31抗体对肿瘤周围血管进行免疫标记并在免疫荧光显微镜下观察并统计定量。结果显示(图5e),与对照组相比,谷氨酰胺酶抑制剂gi-003或贝伐单抗单独使用时,对肿瘤血管生成无明显抑制作用(均<5%),而二者联用时对肿瘤血管生成的抑制高达70%以上。说明该药物组合物腹腔注射入小鼠时有显著加成效果,对肿瘤起到积极的治疗作用。
7.5、对小鼠体内植入的人源肿瘤生长的抑制作用
将源自于乳腺癌病人的肿瘤组织约8mm3的小块植入严重联合免疫缺陷小鼠(scidmice)的乳腺,或将源自于肺癌病人的肿瘤组织约8mm3的小块植入严重联合免疫缺陷小鼠(scidmice)的皮下,建立相应的人源肿瘤小鼠模型(pdxmodel),2-6周后,肿瘤长到约120mm3,开始用谷氨酰胺酶抑制剂或肿瘤血管生成抑制剂贝伐单抗,或者二者联合使用处理肿瘤,其方法是:每隔两天腹腔注射200μl的谷氨酰胺酶抑制剂(bptes:2mg/kg小鼠体重,gi-003:0.5mg/kg小鼠体重),贝伐单抗(2mg/kg小鼠体重),或者二者联合,持续处理4-6周。对照动物用同样的方法注射不含药物的同样助溶载体。在此期间,毎周测定肿瘤大小,计算肿瘤的体积。图6显示的是人源肺癌肿瘤株hlc-3(图6a)和人源乳腺癌肿瘤株hbc-2(图6b)的结果,所选用谷氨酰胺酶抑制剂分别为化合物gi-003和bptes。与对照组相比,谷氨酰胺酶抑制剂或贝伐单抗单独使用时,对肿瘤生长的抑制均低于15%,而二者联用时对肿瘤生长的抑制高达90%以上。与7.4中实验类似,药物处理期结束后,将肿瘤及周边组织取出,切片后进行免疫组化研究。使用抗cd31抗体对肿瘤周围血管进行免疫标记并在免疫荧光显微镜下观察并统计定量。结果显示(图6c和6d),与对照组相比,谷氨酰胺酶抑制剂gi-003/bptes或贝伐单抗单独使用时,对肿瘤血管生成无明显抑制作用(均小于12%),而二者联用时对肿瘤血管生成的抑制高达75%以上。以上均说明该药物组合物腹腔注射入小鼠时有显著加成效果。对肿瘤起到积极的治疗作用。
【实施例8】本发明涉及的药物组合物对小鼠体内肿瘤转移的抑制作用
使用类似于7.5中所述的人源肿瘤小鼠模型(pdxmodel),约6周后,肿瘤长到约100mm3,开始用谷氨酰胺酶抑制剂(gi-003,bptes或cb-839)或肿瘤血管生成抑制剂(贝伐单抗(bevacizumab)或舒尼替尼(sunitinib,20mg/kg小鼠体重))或者二者联合使用处理肿瘤,处理方法与7.5中所述相同。持续处理4-6周后,将小鼠肺部取出,切片,进行组化研究,观察人源肿瘤在小鼠肺部的转移。高转移的人源乳腺癌肿瘤株hbc-5在小鼠肺部的转移的结果见表1-3。由表1和表2可见,与对照组相比,谷氨酰胺酶抑制剂(gi-003或bptes)或贝伐单抗单独使用时,对肿瘤在小鼠肺部的转移无明显抑制作用,而二者联用时对肿瘤在小鼠肺部的转移的抑制高达85%以上。而谷氨酰胺酶抑制剂cb-839和肿瘤血管生成抑制剂舒尼替尼的药物组合也有类似的效果(表3)。
表1:人源乳腺癌肿瘤株hbc-5在小鼠肺部的转移
表2:人源乳腺癌肿瘤株hbc-5在小鼠肺部的转移
表3:人源乳腺癌肿瘤株hbc-5在小鼠肺部的转移
参考文献:
1.deberardinisr.j.,lumj.j.,hatzivassilioug.&thompsonc.b.(2008)thebiologyofcancer:metabolicreprogrammingfuelscellgrowthandproliferation.cellmetab.7:11-20.
2.hsupp,sabatinidm(2008)cancercellmetabolism:warburgandbeyond.cell134:703-707.
3.wised.r.,thompsonc.b.(2010)glutamineaddiction:anewtherapeutictargetincancer.trendsbiochem.sci.35:427-433.
4.shapirora,haserwg,curthoysnp(1985)theorientationofphosphate-dependentglutaminasaontheinnermembraneofratrenalmitochondria.archbiochembioph243:1-7.
5.w.lu,h.pelicano,p.huang(2010)cancermetabolism:isglutaminesweeterthanglucose.cancercell18:199-200.
6.soubaww.(1993)glutamineandcancer.annsurg.218:715-728.
7.loboc.,ruiz-bellidom.,aledoj.,marquezj.,
8.uspatent:us6451828b1:selectiveinhibitionofglutaminasebybis-thiadiazoles.
9.mackeyjr,kerbelrs,gelmonka,mcleoddm,chiask,raysond,vermas,collinsll,patersonah,robidouxa,etal.(2012)controllingangiogenesisinbreastcancer:asystematicreviewofanti-angiogenictrials.cancertreatrev.38:673-688.
10.ferrara,n.,andkerbel,r.s.(2005)angiogenesisasatherapeutictarget.nature438:967–974.
11.carmeliet,p.,andjain,r.k.(2011)molecularmechanismsandclinicalapplicationsofangiogenesis.nature473:298-307.
12.bergers,g.,andhanahan,d.(2008)modesofresistancetoanti-angiogenictherapy.nat.rev.cancer8:592-603.
13.grothey,a.,andgalanis,e.(2009)targetingangiogenesis:progresswithanti-vegftreatmentwithlargemolecules.nat.rev.clin.oncol.6:507-518.
14.chemistryofheterocycliccompounds(newyork,ny,unitedstates),1996,vol.32,no.1:30-34.
- 还没有人留言评论。精彩留言会获得点赞!