结晶药物产品的制备的制作方法
本发明涉及n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其比表面积(ssa)范围为约8至约16m2/g、优选约10至约15m2/g,和制备所述颗粒的方法。
发明背景
化合物n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)及其制备已在wo2011/051540中公开。化合物(i)是强效的雄激素受体(ar)调节剂,其可用于治疗癌症,特别是ar依赖性癌症如前列腺癌和其它需要ar拮抗作用的疾病。化合物(i)以下结构表示:
由于吡唑环的氢原子可以在1-和2-位之间存在互变异构平衡,因此技术人员确定的是如本文提及的上述结构和化学名称“n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)”包括化合物(i)的互变异构体,即n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-3-(1-羟基乙基)-1h-吡唑-5-甲酰胺。
化合物(i)水溶解性差。溶解性差的化合物通常口服生物利用度低。通过微粉化常规地尝试提高溶解性能差的药物的生物利用度。微粉化,即将粒径减小到仅几微米的范围,通常通过增加的比表面积(ssa)而增加溶解性差药物的溶出速率。然而,微粉化颗粒通常具有差的流动性和分散性,导致后续药物加工中的缺陷。
wo2016/120530公开了化合物(i)的稳定结晶形式和通过从乙腈和水的混合物中结晶而制备其的方法。该方法产生具有锐边的小的不规则颗粒。这样的颗粒就药物加工目的而言也不是最佳的,例如由于粉末的流动性差或分离麻烦的原因。因此,需要化合物(i)的结晶颗粒,其更好地适合于药物加工。
发明概述
现已发现,化合物(i)可以作为结晶颗粒从结晶溶剂中获得,其具有更好的后续药物加工性能。在一个方面,所获得的颗粒具有一致且相对高的比表面积(ssa),其在8–16m2/g的范围内,优选10–15m2/g的范围内,大的体积中值直径,例如在100-1000μm的范围内,和窄的粒度分布。在另一方面,颗粒具有圆形颗粒形状。具有圆形颗粒形状的颗粒的特征在于基本上没有锐边。本发明的颗粒易于分离、自由流动并且表现出降低的粘性。此外,发现即使颗粒的体积中值直径减小到10-100μm的范围(例如通过研磨进行),在约8至约16m2/g、优选约约10至约15m2/g的范围内的颗粒比表面积(ssa)也没有显著变化。这确定了一致的生物利用度,而不受粒径如何变化的影响。
因此,根据本发明的颗粒特别适用于药物加工。
因此,根据一个方面,本发明提供了n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其具有在约8至约16m2/g、优选约10至约15m2/g的范围内的比表面积(ssa)。
根据另一方面,本发明提供了n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其具有在约8至约16m2/g、优选约10至约15m2/g的范围内的比表面积(ssa)和≥10μm、优选≥15μm、更优选≥20μm的体积中值直径(dv50)。
根据另一方面,本发明提供了n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其具有在约8至约16m2/g、优选约10至约15m2/g的范围内的比表面积(ssa)和10–1000μm、优选15–800μm、更优选20–750μm的体积中值直径(dv50)。
根据另一方面,本发明提供了n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其具有在约8至约16m2/g、优选约10至约15m2/g的范围内的比表面积(ssa)和100–1000μm、优选120–800μm、更优选150–750μm的体积中值直径(dv50)。根据本发明上述实施方案的一个具体方面,所述结晶颗粒具有圆形颗粒形状。
根据又一方面,本发明提供了n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其具有100–1000μm、优选120–800μm、更优选150–750μm的体积中值直径(dv50)和圆形颗粒形状。
根据又一方面,本发明提供了包含n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)作为活性成分的药物剂型,其中活性成分是根据任何上述实施方案的结晶颗粒形式。
根据又一方面,本发明提供了药物剂型,其中活性成分由n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒来制备,其具有100–1000μm的体积中值直径(dv50)和圆形颗粒形状,例如通过研磨所述颗粒以提供10–100μm的体积中值直径(dv50)。
根据又一方面,本发明提供了药物剂型,其中活性成分由n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒来制备,其具有在约8至约16m2/g、优选约10至约15m2/g的范围内的比表面积(ssa)、100–1000μm的体积中值直径(dv50)和圆形颗粒形状,例如通过研磨所述颗粒以提供10–100μm的体积中值直径(dv50)。
根据又一方面,本发明提供了制备n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒的方法,所述方法包括以下步骤
a)提供在包含乙醇和水的溶剂中的n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i),其中水的量为溶剂重量的35–60%、优选40–58%、更优选42–55%;
b)将混合物加热至约回流温度直至n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)溶解;
c)在至少3小时、优选约4至约8小时的时间期间将混合物冷却至约20–35℃,任选地加入晶种;
d)在至少1小时、优选约2至约10小时时间期间加入水,使得在步骤d)之后,溶剂中水的量为溶剂重量的55–80%、优选58–78%、更优选60–75%,所述加入水任选地与步骤c)同时进行;
e)分离沉淀物。
根据又一方面,本发明提供了n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒,其具有100–1000μm、优选120–800μm、更优选150–750μm的体积中值直径(dv50)并具有圆形颗粒形状,所述颗粒可通过包括以下步骤的方法获得
a)提供在包含乙醇和水的溶剂中的n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i),其中水的量为溶剂重量的35–60%、优选40–58%、更优选42–55%;
b)将混合物加热至约回流温度直至n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)溶解;
c)在至少3小时、优选约4至约8小时的时间期间将混合物冷却至约20–35℃,任选地加入晶种;
d)在至少1小时、优选约2至约10小时的时间期间加入水,使得在步骤d)之后,溶剂中水的量为所述溶剂重量的55–80%、优选58–78%、更优选60–75%,所述加入水任选地与步骤c)同时进行;
和
e)分离沉淀物。
根据一个具体实施方案,通过上述方法可获得的颗粒的比表面积(ssa)范围为约8至约16m2/g,优选约10至约15m2/g。
附图简述
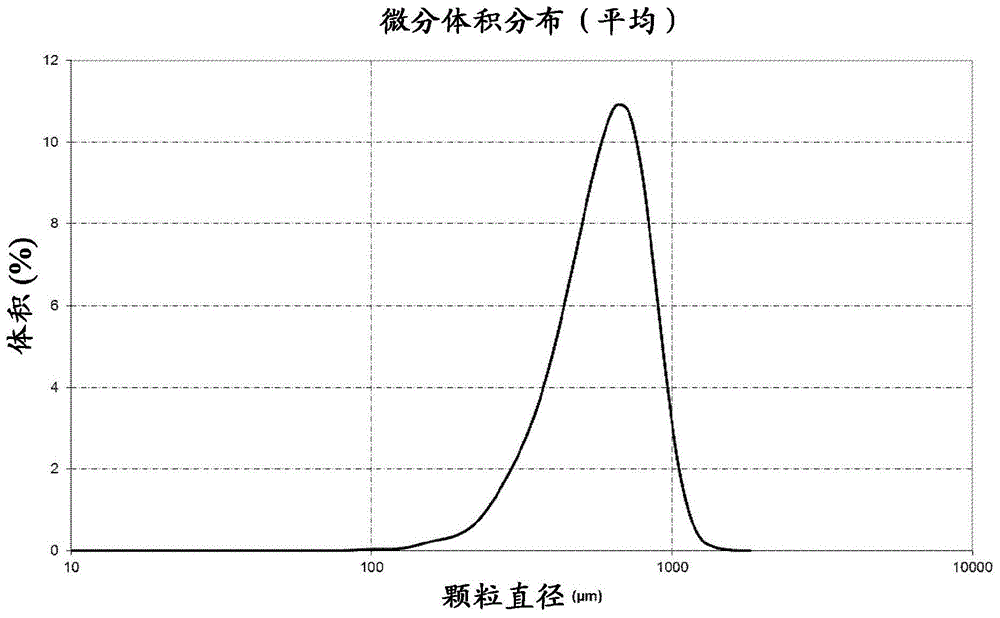
图1显示了通过激光衍射分析的根据本发明制备的化合物(i)的结晶颗粒的粒度分布。
图2显示了根据本发明制备的化合物(i)的结晶颗粒的扫描电子显微镜图像(50倍放大,条长500μm)。
图3(参比)显示了根据wo2016/120530的实施例1制备的化合物(i)的颗粒的扫描电子显微镜图像(500倍放大)。
发明详述
本文所用的术语“具有圆形颗粒形状的颗粒”是指根据本发明的颗粒,其具有基本上球形、椭圆形或马铃薯样的几何形状,其具有基本上没有尖锐或粗糙的边缘的曲面,当在扫描电子显微镜下检查颗粒时,特别是放大50-100倍时,这种几何形状和表面是一致的和明显的。根据本发明的圆形颗粒的特征还在于具有高于0.8、优选高于0.82的平均纵横比和/或高于0.89、优选高于0.9的平均hs(高灵敏度)圆形度。
如本文所用,术语“纵横比”是指颗粒的最短尺寸与最长尺寸的比率,并且在0至1的范围内。
如本文所用,术语“高灵敏度(hs)圆形度”是指等于圆形度的平方的参数,其中圆形度等于:等于颗粒的投影面积的圆的周缘与颗粒的实际周缘(周长)的比率。因此,高灵敏度(hs)圆形度计算为(4π×面积)/(周长2)。
颗粒的平均纵横比和平均高灵敏度(hs)圆形度可以通过基于光学显微镜的方法在干分散体上测定,如morphologig3tm粒度和颗粒形状分析仪(malverninstruments)。可以通过使用morphologig3tm集成干粉分散器(malverninstruments)制备样品,例如使用7mm3的样品量和1.0巴的分散压力。自动图像分析适合在没有过滤器的情况下进行。施加的放大率取决于所分析的粉末的粒度,通常为10倍。
如本文所用的术语“n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒”是指化合物(i)的颗粒,其中化合物(i)至少部分为结晶形式,包括微晶形式。例如,该术语包括化合物(i)的颗粒,其中化合物(i)至少部分为在wo2016/120530中公开的结晶形式i。结晶形式i的x射线粉末衍射(xrpd)图案在约8.5、10.4、16.6、16.9和24.3度2-θ处具有特征峰。因此,该术语包括在约8.5、10.4、16.6、16.9和24.3度2-θ处显示xrpd特征峰的颗粒。
化合物(i)的结晶颗粒的粒度分布可以通过激光衍射分析,例如使用配备有tornado干粉体系的beckmancoulterls13320激光衍射粒度分析仪,使用空气作为分散介质,测定压力为24”h2o±2”h2o,样品量10毫升,就浊度而言系统控制目标为5%并应用弗劳恩霍夫(fraunhofer)光学模型。
考虑的参数是颗粒的第10、50和90百分位的体积直径(μm),分别表示为dv10、dv50和dv90,其通过假定颗粒具有等于球形的几何形状来确定。
化合物(i)的结晶颗粒的比表面积(ssa)可以使用基于brunauer、emmett和teller(bet)理论的三点氮吸附技术进行分析,例如使用tristar3000自动气体吸附分析仪(micromeritics,inc.)。将样品适当地在40℃下真空干燥20小时。体积法可以在0.1-0.3p/p0的相对压力范围内使用。
本发明提供了一种制备n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒的方法,所述方法包括以下步骤:
a)提供在包含乙醇和水的溶剂中的n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i),其中水的量为溶剂重量的35–60%、优选40–58%、更优选42–55%;
b)将混合物加热至约回流温度直至n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)溶解;
c)在至少3小时、优选约4至约8小时的时间期间将混合物冷却至约20–35℃,任选地加入晶种;
d)在至少1小时、优选约2至约10小时的时间期间加入水,使得在步骤d)之后,溶剂中水的量为溶剂重量的55–80%、优选58–78%、更优选60–75%,所述加入水任选地与步骤c)同时进行;和
e)分离沉淀物。
步骤a)中使用的溶剂通常包含乙醇和水。步骤a)的溶剂中水的量为溶剂重量的约35–60%、优选40–58%、更优选42–55%。优选地,溶剂基本上由乙醇和水组成。例如,步骤a)的溶剂包含溶剂重量的35–60%的水和40–65%的乙醇、优选40–58%的水和42–60%的乙醇、更优选42–55%的水和45–58%的乙醇。根据一个实施方案,步骤a)的溶剂包含溶剂重量的45–52%的水和48–55%的乙醇。根据另一个实施方案,步骤a)的溶剂包含溶剂重量的48–55%的水和45–52%的乙醇。
步骤a)中使用的化合物(i)的量合适地为溶剂重量的约1–20%、优选约5-15%、例如6–12%。例如,在合适的反应器中,在1500–3800kg乙醇-水溶剂中提供150-250kg化合物(i)。然后将混合物在搅拌下加热,合适地加热至约回流温度,例如加热至约65-85℃,直至化合物(i)溶解。在步骤c)中,然后将混合物缓慢冷却至20-35℃,同时温和搅拌,通常搅拌速度小于80rpm。冷却至少进行3小时、优选进行约4至约8小时,任选地加入化合物(i)的晶体作为晶种。加入晶种适合在从约75℃开始的温度进行,并且任选地在较低的温度进行。例如,当混合物的温度为约50-70℃时,可以一次或多次加入晶种。加入晶种用的晶体的量通常小于最初提供给反应器的化合物(i)重量的0.5%。作为晶种的化合物(i)的晶体可以例如使用wo2016/120530中描述的方法制备。
在步骤d)中,向混合物中缓慢加入更多的水,使得在加入水后,溶剂中水的量为溶剂重量的55–80%、优选58–78%、更优选60–75%。优选地,溶剂基本上由乙醇和水组成。例如,步骤d)后的溶剂包含溶剂重量的55–80%的水和20–45%的乙醇、优选58–78%的水和22–42%的乙醇、更优选60–75%的水和25–40%的乙醇。
根据一个实施方案,步骤d)之后的溶剂包含溶剂重量的60–65%的水和35–40%的乙醇。根据另一个实施方案,步骤d)之后的溶剂包含溶剂重量的65–70%的水和30–35%的乙醇。根据又一个实施方案,步骤d)之后的溶剂包含溶剂重量的70–75%的水和25–30%的乙醇。
根据另一个实施方案,步骤a)的溶剂包含溶剂重量的48–55%的水和45–52%的乙醇,且步骤d)后的溶剂包含溶剂重量的60–65%的水和35–40%的乙醇。根据另一个实施方案,步骤a)的溶剂包含溶剂重量的45–52%的水和48–55%的乙醇,且步骤d)后的溶剂包含溶剂重量的70–75%的水和25–30%的乙醇。
水的加入在至少1小时、优选约2至约10小时、例如约6至约10小时的时间期间进行。在加水期间将该混合物温和搅拌,通常搅拌速度小于80rpm。在加水期间,将混合物的温度适当地保持在约20–35℃。
或者,步骤c)和d)可以同时进行。在该实施方案中,在冷却步骤期间加入水。可以在将混合物冷却至约20-35℃的同时进行如上所述的加水程序,包括任选加入晶种。同时进行的冷却和加水适合在至少3小时内进行、优选在4-10小时时间期间进行。
在步骤d)之后,可以在至少1小时,例如1-3小时时间期间将混合物进一步冷却、至优选至少10-30℃,例如10-20℃。冷却后,适当搅拌混合物直至沉淀完成。沉淀的结晶颗粒易于分离,例如通过离心、然后用水和/或乙醇洗涤。分离的沉淀物可以在减压下、例如在真空下、在至少30℃例如40-60℃的温度干燥一段时间,以完成干燥。
通过上述方法获得的颗粒是结晶的,通常具有圆形颗粒形状,并且显现出的比表面积(ssa)通常为约8至约16m2/g,更通常为约10至约15m2/g。获得的颗粒通常具有100–1000μm、优选120–800μm、更优选150–750μm、特别是180–700μm、例如200–650μm的体积中值直径(dv50)。dv10通常大于约50μm、优选大于约60μm、更优选大于约70μm、特别是80–500μm、例如100–400μm。dv90通常低于2000μm、优选低于1500μm、更优选低于1400μm、特别是300–1300μm、例如400–1200μm。
此外,80体积-%的颗粒通常为50–2000μm、优选60–1500μm、更优选70–1400μm、特别是80–1300μm、例如100–1200μm。
通过上述方法获得的圆形颗粒的特征通常在于平均纵横比高于0.8和/或平均高灵敏度(hs)圆形度高于0.89。更典型地,圆形颗粒的特征在于平均纵横比高于0.8且平均高灵敏度(hs)圆形度高于0.89。更典型地,圆形颗粒的特征在于平均纵横比高于0.82且平均高灵敏度(hs)圆形度高于0.9。
由于通过上述方法获得的颗粒具有大的体积中值直径、窄的粒度分布和圆形颗粒形状,其特征在于基本上没有锐边,它们易于分离,自由流动并且表现出降低的粘性。通过上述方法获得的圆形颗粒的比表面积(ssa)为约8至约16m2/g、优选约10至约15m2/g,并且即使减小在颗粒的体积中值直径(dv50)的情况下、例如通过碾磨或其他合适方式减小到10-100μm的范围,比表面积也没有显著变化。这确定了一致的生物利用度,而不受粒径如何变化的影响。因此,如果压片物质的均匀性更高(如需要),则可以将圆形颗粒研磨成具有例如10-100μm、优选15-95μm、通常20-90μm的范围内的dv50的粒度,所述颗粒非常适合用于制备口服施用的药物剂型如片剂。
因此,通过本发明的方法获得的化合物(i)的结晶圆形颗粒可以以原样或以研磨的形式与本领域已知的赋形剂一起用于药物剂型的制备,如片剂、胶囊或粉末。
通过以下实施例进一步说明本发明。
实施例1.n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒的制备
将颗粒状硼氢化钠(15kg)和etoh(1370kg)置于6.3m3的搪瓷反应容器中。通过在22℃下搅拌30分钟溶解该混合物。将(s)-3-乙酰基-n-(1-(3-(3-氯-4-氰基-苯基)-1h-吡唑-1-基)丙烷-2-基)-1h-吡唑-5-甲酰胺(225kg)加入反应容器中。然后将该混合物在22℃搅拌4小时以完成反应。然后用hcl水溶液将混合物的ph调节至酸性。然后加入水(800kg),并通过加入在水中的naoh将混合物的ph设定为7.0±1.0。将该混合物温至65℃,然后转移至6.3m3夹套钢反应容器中。将该混合物温至78℃以溶解混合物。将该溶液在氮气氛下冷却至64℃。向该溶液在64℃在温和搅拌下加入晶种。然后将该溶液在8h期间在温和搅拌下冷却至30℃。然后在7-10h期间在30℃温和搅拌下加入水(2600kg)。将该混合物在2h期间在温和搅拌下冷却至20℃,然后再搅拌1h。通过离心分离沉淀的产物,用水洗涤,并在40-60℃下真空干燥,得到214kg圆形颗粒形状的结晶颗粒。
实施例2.n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒的制备
将水(450kg)、etoh(920kg)和(n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)丙烷-2-基)-3-(1-羟基乙基)-1h-吡唑-5-甲酰胺(215kg)置于有100kg清洗(rinse)etoh的6.3m3钢反应容器中。通过加热至75℃将该混合物溶解。加入活性炭sxultra(11kg)和硅藻土(21kg),随后在78℃搅拌1h。将该混合物在氮气氛下冷却至75℃,并过滤。将滤液转移至6.3m3夹套钢反应容器。将碳/硅藻土饼用水(970kg)和etoh(345kg)的温(75℃)混合物洗涤。洗涤液也加入反应容器中。将该溶液在78℃搅拌30min,然后冷却至70℃。在余下的操作中保持温和搅拌。将该溶液在70℃加入晶种,然后在4h期间冷却至30±5℃。然后在6h期间在30±5℃加入水(840kg)。在2h期间将该混合物冷却至20℃,然后再搅拌1h。通过离心分离沉淀的产物,用etoh洗涤,并在40-60℃真空干燥,得到190kg圆形颗粒形状的结晶颗粒。
实施例3.n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)丙烷-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(i)的结晶颗粒的制备
将水(1400kg)、etoh(1215kg)和(n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)丙烷-2-基)-3-(1-羟基乙基)-1h-吡唑-5-甲酰胺(210kg)置于6.3m3钢反应容器中。通过加热至75℃将该混合物溶解。加入活性炭sxultra(11kg)和硅藻土(21kg),随后搅拌1h。然后将该混合物热过滤。将滤液转移至6.3m3夹套钢反应容器中。将碳/硅藻土饼用etoh(170kg)洗涤。将洗涤液也加入反应容器中。温度调节至70℃。将该溶液在70℃加入晶种,然后冷却至60℃。然后在4小时内将该混合物冷却至30℃,并同时加入水(1050kg)。将混合物再搅拌30分钟。离心分离沉淀的产物,用水洗涤,并在70℃真空干燥,得到190kg圆形颗粒形状的结晶颗粒。
实施例4.粒度分布的确定
通过激光衍射确定根据本发明制备的化合物(i)的结晶圆形颗粒的粒度分布。使用配备有tornado干粉系统的beckmancoulterls13320激光衍射粒度分析仪进行测定,使用空气作为分散介质,测定压力为24”h2o±2”h2o,样品量为10ml,就浊度而言系统控制目标为5%并应用弗劳恩霍夫光学模型。粒度分析的结果如图1所示。根据分析,颗粒的dv10值为359μm,dv50为632μm,dv90为925μm。
实施例5.通过扫描电子显微镜(sem)图像表征颗粒
通过扫描电子显微镜成像表征根据本发明制备的化合物(i)的结晶圆形颗粒。sem图显示在图2中(50倍放大,条长500μm)。作为比较,根据wo2016/120530的实施例1制备的颗粒的sem图像在图3中显示(500倍放大,条长30μm)。根据本发明制备的颗粒呈现圆形颗粒形状,具有窄的粒度分布,而根据wo2016/120530制备的颗粒小且不规则,具有锐边。
实施例6.颗粒比表面积(ssa)的确定
确定两批(a和b)根据本发明制备的化合物(i)的结晶圆形颗粒的比表面积(ssa)和粒度分布(psd)。这两批颗粒然后被研磨,接着确定ssa和psd。结果显示在表1和2中。结果表明,即使将颗粒研磨至减小的粒度,颗粒的比表面积(ssa)也没有显著变化。
表1
表2
使用基于brunauer、emmett和teller(bet)理论的三点氮吸附技术使用tristar3000自动气体吸附分析仪(micromeritics,inc.)测定比表面积。将样品在40℃真空干燥20小时。该容量法在0.1-0.3p/p0的相对压力范围内应用。
- 还没有人留言评论。精彩留言会获得点赞!