包含吲哚萘并吡喃的制品的制作方法
本发明涉及制品,如光学制品,其包含某些吲哚萘并吡喃化合物,特别是光致变色吲哚萘并吡喃。
背景技术:
光致变色化合物响应于电磁辐射(例如,“光化辐射”)的某些波长而经受从一种状态(或形式)至另一种状态的转变。各状态具有特征吸收光谱。例如,许多光致变色化合物在暴露于光化辐射时从未活化(例如,脱色或基本上无色)的第一状态转变为活化(例如,着色或变暗)的第二状态。当光化辐射被移除时,光致变色化合物可逆地从第二种活化状态转变回第一种未活化状态。
光致变色化合物可以关于各种特性来表征,诸如但不限于:褪色速率;光密度变化(δod);饱和时光密度变化(δod);灵敏度(δod/分钟);光致变色化合物吸收活化光致变色化合物所需要的辐射的效率(色度);以及二向色性特性,诸如在光致变色-二向色性化合物的情况下,其可以关于吸收比(ar)值被量化。光密度变化测量从未活化状态至活化状态的变化。
一些光致变色化合物具有双峰吸收曲线,其具有强度大于吸收带“b”(后文称为“b带”)的吸收带“a”(后文称为“a带”)。a波段的吸收通常发生在420-500纳米波长范围内,而b波段的吸收发生在活化可见光谱的500-650纳米波长范围内。
黄色着色的萘并吡喃在活化(即着色)状态下具有高b*值,但这些化合物在耐久性方面具有固有的弱点,并且经常以比茚并稠合萘并吡喃家族的紫色或蓝色光致变色化合物快得多的速率降解。宽带吸收茚并稠合化合物已经被用于克服黄色萘并吡喃的局限,但在活化(即着色)状态下呈黄色(高b*)的茚并稠合萘并吡喃仍然难以获得。
蓝光对健康的有害影响,如白内障、黄斑变性和昼夜节律的破坏,已得到充分记录。已经生产了许多固定色调光学产品,以帮助阻挡来自室内光源(如来自荧光照明)和室外暴露在具有高水平蓝光存在的阳光下的有害蓝光。此类常规光学产品通常使用静态黄色染料或色调作为蓝光阻挡剂。然而,包含这些静态黄色染料的此类光学产品的静态黄色在美学上是不希望的,特别是当在室内配戴时。此外,当在户外配戴时,此类光学产品不能增加对阳光中存在的蓝光的防护。也就是说,依赖静态黄色染料或色调来阻挡蓝光的常规光学制品在室内和室外都具有黄色,并且在室内和室外都提供相同水平的蓝光阻挡。
因此,希望提供一种制品,如光学制品,其在室内环境中基本上是透明的并且稍微阻挡蓝光,但在室外环境中暴露于阳光下时会变色,并变得更具有蓝光阻挡性。
技术实现要素:
本发明涉及一种制品,其响应于光化辐射从第一状态转变为第二状态。该制品包含吲哚萘并吡喃,其中在第一状态下该制品展现出大于80%的透射率百分比,并且在第二状态下该制品展现出在35%与75%之间的透射率百分比,以及小于20的τsb值。
在权利要求书中具体指出了表征本发明的特征,将其附在本披露内容中并使其构成本披露内容的一部分。从以下详细说明中将更全面地理解本发明的这些和其他特征、其操作优点以及通过其使用所获得的具体目的,在以下详细说明中展示并描述了本发明的非限制性实施例。
附图说明
图1说明了用于制备可用于制备本发明制品的光致变色吲哚萘并吡喃化合物的示例性方法的一般方案,方案1。
具体实施方式
如本文所使用的,冠词“一个/种(a/an)”、以及“所述(the)”包括复数指示物,除非另有清楚地且明确地限于一个指示物。
如本文所使用的,术语“包括(includes)”与“包含/包括(comprises)”同义。
除非另有指明,否则本文所披露的所有范围或比率应理解为涵盖其中所包含的任何和所有子范围或子比率。例如,叙述的范围或比率“1至10”应视为包括在最小值1与最大值10之间(并且包含端点)的任何和所有子范围;即,以最小值1或更大值开始并且以最大值10或更小值结束的所有子范围或子比率,诸如但不限于,1至6.1、3.5至7.8、以及5.5至10。
如本文所使用的,除非另有指明,连接基团(诸如二价连接基团)的从左至右表示包括其他合适的取向,诸如但不限于从右至左取向。出于非限制性说明的目的,二价连接基团
除了在操作实例中、或在另有指明的情况下,在说明书和权利要求书中所使用的表示成分、反应条件等的量的所有数字应理解为在所有情况下由术语“约(about)”修饰。“约”意指所述值的正或负百分之二十五,诸如所述值的正或负百分之十。然而,不应将其视为对等效原则下的值的任何分析的限制。
如本文所使用的,聚合物的分子量值,诸如重均分子量(mw)和数均分子量(mn)通过凝胶渗透色谱法使用合适的标准物(诸如聚苯乙烯标准物)来确定。
如本文所使用的,术语“聚合物”意指均聚物(例如,由单一单体种类所制备的)、共聚物(例如,由至少两种单体种类所制备的)、以及接枝聚合物。
如本文所使用的,术语“(甲基)丙烯酸酯((meth)acrylate)”和类似术语,诸如“(甲基)丙烯酸酯((meth)acrylicacidester)”,意指丙烯酸和甲基丙烯酸的衍生物,包括丙烯酸酯类、甲基丙烯酸酯类、丙烯酰胺类、甲基丙烯酰胺类、丙烯酸和甲基丙烯酸。如本文所使用的,术语“(甲基)丙烯酸((meth)acrylicacid)”意指甲基丙烯酸和/或丙烯酸。
在一些实施例中,本发明的光致变色化合物在本文也称为光致变色-二向色性化合物(诸如,当它们包括一个或多个含介晶的基团诸如l1时)。
如本文所描述的,本发明的光致变色化合物(包括但不限于由式(i)和式(ia)表示的光致变色化合物)在各情况下可以任选地进一步包括由此类化合物的合成产生的一种或多种副产物。
如本文所使用的,术语“光致变色(的)(photochromic)”和类似术语(诸如“光致变色化合物”)意指具有响应于至少光化辐射的吸收而变化的至少可见辐射的吸收光谱。进一步地,如本文所使用的,术语“光致变色材料”意指适于显示光致变色特性(诸如适于具有响应于至少光化辐射的吸收而变化的至少可见辐射的吸收光谱)并且包括至少一种光致变色化合物的任何物质。
如本文所使用的,术语“光化辐射”意指能够在材料中引起响应的电磁辐射,诸如但不限于将光致变色材料从一种形式或状态转变至另一种形式或状态,如本文将进一步详细讨论的。
如本文所使用的,术语“二向色性”意指能够比另一种更强地吸收至少透过辐射的两种正交平面偏振分量中的一种。
如本文所使用的,术语“光致变色-二向色性”和类似术语(诸如“光致变色-二向色性化合物”)意指拥有和/或提供光致变色特性(即,具有响应于至少光化辐射而变化的至少可见辐射的吸收光谱)和二向色性特性(即,能够比另一种更强地吸收至少透过辐射的两种正交平面偏振分量中的一种)两者。
如本文所使用的,并且除非另有说明或另有限制,术语“光致变色材料”包括热可逆光致变色材料和化合物以及非热可逆光致变色材料和化合物。如本文所使用的术语“热可逆光致变色化合物/材料”意指能够响应于光化辐射从第一状态(例如“透明状态”)转换至第二状态(例如“着色状态”)、并且响应于热能返回至第一状态的化合物/材料。如本文所使用的术语“非热可逆光致变色化合物/材料”意指能够响应于光化辐射从第一状态(例如“透明状态”)转换至第二状态(例如“着色状态”)、并且响应于与着色状态的吸收基本上相同波长的光化辐射(例如,停止暴露于此光化辐射)返回至第一状态的化合物/材料。
如本文所使用的,为了修饰术语“状态(state)”,术语“第一(first)”和“第二(second)”不旨在指任何特定顺序或时间顺序,而是指两种不同的条件或特性。出于非限制性说明的目的,光致变色化合物的第一状态和第二状态可以在至少一种光学特性方面不同,诸如但不限于可见辐射和/或uv辐射的吸收。因此,根据本文所披露的各种非限制性实施例,本发明的光致变色化合物可以在第一和第二状态中的每一个中具有不同的吸收光谱。例如,虽然本文没有限制,本发明的光致变色化合物可以在第一状态下是透明的并且在第二状态下是着色的。替代性地,本发明的光致变色化合物可以在第一状态下具有第一颜色并且在第二状态下具有第二颜色。
如本文所使用的,术语“光学(的)(optical)”意指涉及光和/或视觉或者与光和/或视觉相关。例如,根据本文所披露的各种非限制性实施例,光学制品或元件或器件可以选自眼科制品、元件和器件;显示制品、元件和器件;窗;镜;或有源和无源液晶盒制品、元件和器件。
如本文所使用的,术语“眼科(的)(ophthalmic)”意指涉及眼睛和视觉或者与眼睛和视觉相关。眼科制品或元件的非限制性实例包括矫正和非矫正镜片(其包括单视或多视镜片,多视镜片可以是分段的或非分段的多视镜片(诸如但不限于双焦镜片、三焦镜片和渐进镜片)、以及用于矫正、保护或增强(美容或其他)视觉的其他元件(其包括但不限于接触镜片、眼内镜片、放大镜片、以及保护性镜片或护目镜)。
如本文所使用的,术语“显示(display)”意指呈字、数字、符号、设计或图的信息的可见的或机器可读的表示。显示元件的非限制性实例包括屏幕、监视器、以及安全元件,诸如安全标记。
如本文所使用的,术语“窗(window)”意指适于允许辐射透射穿过那里的孔。窗的非限制性实例包括汽车和飞机的透明体、挡风玻璃、滤光片、遮光器、以及光学开关。
如本文所使用的,术语“镜(mirror)”意指镜面反射很大部分的入射光的表面。
如本文所使用的,术语“液晶盒(liquidcrystalcell)”是指含有能够被有序化的液晶材料的结构。液晶盒元件的非限制性实例是液晶显示器。
如本文所使用的,术语“形成在…上/在…上形成(formedover)”、“沉积在…上/在…上沉积(depositedover)”、“提供在…上/在…上提供(providedover)”、“施用在…上/在…上施用(appliedover)”、“驻留在…上/在…上驻留(residingover)”或“定位在…上/在…上定位(positionedover)”意指形成、沉积、提供、施用、驻留或定位在上面、但不必然与下面的元件、或下面的元件的表面直接(或邻接)接触。例如,“定位在基底上(positionedover)”的层不排除位于所定位或形成的层与基底之间存在相同或不同组成的一个或多个其他层、涂层、或膜。
如本文所使用的,涉及环位置(诸如但不限于位置-x(例如,位置-3或位置-13)的叙述意指化学化合物(诸如本发明的吲哚萘并吡喃光致变色化合物)在环结构(诸如核心骨架结构)中的特定位置,并且其在本文根据一些实施例通过代表性化学式(诸如但不限于式(i)和/或(ia))的环结构内的数字描绘。
“核心骨架结构”意指至少包含相关式中所描绘的骨架结构的化合物。出于识别经编号的环位置的目的提供核心骨架结构。然而,应理解的是,除非明确地相反示出,否则一种或多种核心骨架结构可以具有键合至核心骨架结构上的一个或多个经编号的环位置的一个或多个原子或一个或多个基团(在相应的式中未具体展示),该一个或多个原子或一个或多个基团可以彼此相同或不同。
本发明的光致变色化合物在本文中是指关于术语“核心骨架结构”,其可以由一个或多个式表示,例如但不限于式(i)和/或(ia)。
本文所涉及、并且除非另有指明,所有文献或部分文献(诸如但不限于公布的专利和专利申请)均视为“通过援引以其全文并入”。
如本文所使用的,“取代(的)”基团的叙述意指包括但不限于烷基、杂环烷基、芳基、和/或杂芳基的基团,其中其至少一个氢已经被除氢以外的基团替代或取代,诸如但不限于烷氧基;卤素基团(例如,f、cl、i、以及br);羟基;硫醇基团;烷硫基;酮基团;醛基团;酯基团;羧酸基团;磷酸基团;磷酸酯基团;磺酸基团;磺酸酯基团;硝基;氰基;烷基(包括芳烷基);烯基;炔基;卤代烷基;全卤代烷基;杂环烷基;芳基(包括烷芳基,包括羟基取代的芳基,诸如苯酚,并且包括多稠环芳基);杂芳基(包括多稠环杂芳基);氨基,诸如-n(r11’)(r12’),其中r11’和r12’各自独立地选自例如氢、烷基、杂环烷基、芳基、或杂芳基;羧酸酯基团;硅氧烷基团;烷氧基硅烷基团;聚硅氧烷基团;酰胺基团;氨基甲酸酯基团;碳酸酯基团;脲基团;聚酯基团;聚醚基团;聚碳酸酯基团;聚氨酯基团;丙烯酸酯基团;甲基丙烯酸酯基团;含氮杂环化合物;或它们的组合,包括如本文进一步描述的那些类别和实例。
“芳基”是指芳族环状单价烃基,并且术语“芳族”是指具有稳定性(由于离域)显著大于假定的定域结构的稳定性的环状共轭烃。芳基的实例包括c6-c14芳基,诸如但不限于苯基、萘基、菲基、以及蒽基。
如本文所使用的,“卤素取代(的)”和相关术语(诸如但不限于卤代烷基、卤代烯基、卤代炔基、卤代芳基和卤代杂芳基)的叙述意指其中其至少一个、且至多且包括所有可用氢基团被卤素基团取代的基团。术语“卤代(的)”包括“全卤代(的)”。如本文所使用的,术语全卤代基团和相关术语(诸如但不限于全卤代烷基、全卤代烯基、全卤代炔基、全卤代芳基或全卤代杂芳基)意指其中其所有可用氢基团被卤素基团取代的基团。例如,全卤代甲基是-cx3;全卤代苯基是-c6x5,其中x表示一个或多个卤代基团,诸如但不限于f、cl或br。
如本文所使用的,“直链或支链(的)”基团(诸如直链或支链烷基)的叙述在本文被理解为包括:直链(或“直链(straightchain)”)基团,诸如直链c1-c25烷基;以及适当支化的基团,诸如支链c3-c25烷基。
本文所使用的术语“烷基”意指直链或支链的、环状或非环状的c1-c25烷基。直链或支链烷基可以包括c1-c25烷基,诸如c1-c20烷基,诸如c2-c10烷基,诸如c1-c12烷基,诸如c1-c6烷基。本发明的各种烷基可以选自其的烷基的实例包括但不限于本文进一步列举的那些。烷基可以包括“环烷基”基团。本文所使用的术语“环烷基”意指适当环状的基团,诸如但不限于c3-c12环烷基(包括但不限于环状c5-c7烷基、或环状c3-c10烷基)基团。环烷基的实例包括但不限于本文进一步列举的那些。本文所使用的术语“环烷基”还包括:桥环多环烷基(polycycloalkyl)基团(或桥环多环(polycyclic)烷基),诸如但不限于双环[2.2.1]庚基(或降冰片基)和双环[2.2.2]辛基;以及稠环多环烷基(polycycloalkyl)基团(或稠环多环(polycyclic)烷基),诸如但不限于八氢-1h-茚基、以及十氢萘基。
本文所使用的术语“杂环烷基”意指适当环状的基团,诸如但不限于c2-c12杂环烷基,诸如c5-c7杂环烷基、诸如c2-c10杂环烷基,并且其在环状环中具有至少一个杂原子,诸如但不限于o、s、n、p、以及它们的组合。杂环烷基的实例包括但不限于咪唑基、四氢呋喃基、四氢吡喃基和哌啶基。本文所使用的术语“杂环烷基”还包括:桥环多环杂环烷基,诸如但不限于7-氧杂双环[2.2.1]庚烷基;以及稠环多环杂环烷基,诸如但不限于八氢环戊并[b]吡喃基和八氢-1h-异色烯基。
如本文所使用的,术语“杂芳基”包括但不限于c3-c18杂芳基,例如但不限于c3-c10杂芳基(包括稠环多环杂芳基)并且意指在芳环中,或者在稠环多环杂芳基的情况下在至少一个芳环中具有至少一个杂原子的芳基。杂芳基的实例包括但不限于呋喃基、吡喃基、吡啶基、异喹啉、以及嘧啶基。
如本文所使用的,术语“稠环多环-芳基-烷基”和类似术语,如稠环多环-烷基-芳基、稠环多环-芳基-烷基和稠环多环-烷基-芳基意指稠环多环基团,其包括稠合在一起形成稠环结构的至少一个芳基环和至少一个环烷基环。出于非限制性说明的目的,稠环多环-芳基-烷基的实例包括但不限于茚基、9h-芴基、环戊萘基和indacenyl。
如本文所使用的,术语“芳烷基”包括但不限于c6-c24芳烷基,例如但不限于c6-c10芳烷基,并且意指被芳基取代的烷基。芳烷基的实例包括但不限于苄基和苯乙基。
代表性的烷基包括但不限于甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、新戊基、己基、庚基、辛基、壬基和癸基。代表性烯基包括但不限于乙烯基、烯丙基和丙烯基。代表性炔基包括但不限于乙炔基、1-丙炔基、2-丙炔基、1-丁炔基、以及2-丁炔基。代表性环烷基包括但不限于环丙基、环丁基、环戊基、环己基、以及环辛基取代基。代表性杂环烷基包括但不限于咪唑基、四氢呋喃基、四氢吡喃基和哌啶基。代表性芳基包括但不限于苯基、萘基、蒽基、菲基、以及并四苯基(包括它们的结构性异构体)。代表性杂芳基包括但不限于呋喃基、吡喃基、吡啶基、异喹啉基、以及嘧啶基。代表性芳烷基包括但不限于苄基和苯乙基。
如本文所使用的,术语“含氮杂环”包括但不限于含氮环,其中含氮环通过环氮键合。含氮杂环的实例包括但不限于环氨基类,诸如吗啉代、哌啶子基、以及吡咯烷子基;以及杂芳族化合物,诸如咪唑、吡咯、吲哚、以及咔唑。
如本文所使用的,“…中的至少一个/种”与“…中的一个/种或多个/种”同义,无论要素是结合地列出还是分离地列出。例如,短语“a、b、以及c中的至少一个/种”和“a、b、或c中的至少一个/种”各自意指a、b、或c中的任意一个/种,或者a、b、或c中的任意两个/种或更多个/种的任意组合。例如,单独的a;或单独的b;或单独的c;或a和b;或a和c;或b和c;或全部的a、b、以及c。
如本文所使用的,“选自(selectedfrom)”与“选自(chosenfrom)”同义,无论要素是结合地列出还是分离地列出。进一步地,短语“选自a、b、以及c”和“选自a、b、或c”各自意指a、b、或c中的任意一个/种,或者a、b、或c中的任意两个/种或更多个/种的任意组合。例如,单独的a;或单独的b;或单独的c;或a和b;或a和c;或b和c;或全部的a、b、以及c。
本发明的讨论可以将某些特征描述为“特别地”或“优选地”在某些限制内(例如,“优选地”、“更优选地”、或“甚至更优选地”在某些限制内)。应理解的是,本发明不限于这些特定或优选的限制,而是涵盖本披露的整个范围。
本发明包括、或基本上由本发明的以下方面以任何组合组成。如前所述,本发明涉及一种制品,其响应于暴露于电磁辐射(例如光化辐射)从第一状态转变为第二状态。在第一状态下,制品展现出大于80%,如大于85%,如大于88%的透射率百分比。在第二状态下,制品展现出在35%与75%之间,如在40%与70%之间,如在45%与65%之间的透射率百分比,以及小于20%,如小于15%,如小于10%的τsb值。例如,在第二状态下,制品可以展现出3.0%至小于20%,如4.0%至小于20%,如5.0%至小于20%的τsb值。术语“透射率百分比”意指如由人眼感知的透射通过透明制品的光谱光的百分比,其如使用ciey值根据cie15:2004比色法,使用d65光源和10°观测器来确定。在本文的说明书和实例中,透射率百分比也可以称为“%t”。τsb值是太阳蓝光透射率,其是380与500纳米之间的光谱透射率的平均值和适当的加权函数的结果。参见iso12311:2013(e),第7.4小节。
例如,本发明的制品可以是光致变色制品,其中第一状态是未活化的、通常无色的状态,例如当制品未暴露于光化辐射时。在该第一状态下的透射率百分比大于80%。在暴露于光化辐射时,光致变色制品转变为第二、活化或着色状态,其中透射率百分比响应于光化辐射而降低。在该第二状态下的透射率百分比降低到范围从35%至75%的值。由于在第二状态下响应于光化辐射的透射率百分比降低,τsb值(即,太阳蓝光透射率)也降低至小于20%。因此,本发明的制品响应于光化辐射从通常透明的第一状态转变为具有增强的蓝光阻挡的着色的第二状态。在移除光化辐射源后,制品可恢复到第一未活化且透明或无色的状态。
此外,本发明的制品可以具有在第一状态下范围从-5至15,如从-5至10,如从-5至5的b*值。此外,本发明的制品可以具有在第一状态下范围从0至15,如从0至10,如从0至5的b*值。如在本文的说明书和权利要求书中所使用的b*值是指根据cie15:2004空间比色法,使用d65光源和10°观测器,使用hunterultrascanpro装置测量的b*值。b*轴代表蓝黄色分量,其中蓝色为负方向并且黄色为正方向。如上刚刚所述的b*值表明本发明的制品在第一状态下通常没有或几乎没有黄色。
如前所述,本发明的制品包含吲哚萘并吡喃,例如包含由下式(i)表示的核心骨架结构的光致变色吲哚萘并吡喃。在本发明的制品中使用的吲哚萘并吡喃可以由下述的一种或多种核心骨架结构表示。
式(i)的核心骨架结构的各可用的经编号的环位置(例如,1、2、5、8、9、10、11、和/或12)可以具有与其共价键合的氢或除氢以外的基团,例如,诸如本文所描述的基团。此类基团的实例描述如下。
对于式(i),r1和r2各自独立地是氢、取代或未取代的烷氧基、取代或未取代的芳氧基、取代或未取代的烷硫基、取代或未取代的芳硫基、取代或未取代的醚、取代或未取代的硫醚、氨基、含氮杂环、取代或未取代的烷基、取代或未取代的芳基、-nhc(o)ra、或-oc(o)ra。ra可以选自其的基团的实例包括取代或未取代的烷基、取代或未取代的芳基、取代或未取代的杂芳基、取代或未取代的烷氧基、取代或未取代的芳氧基、取代或未取代的烷硫基、或者取代或未取代的芳硫基。r4选自氢、取代或未取代的烷基、取代或未取代的杂环烷基、烯丙基、取代或未取代的芳基、或者取代或未取代的杂芳基。例如,r4可以是取代或未取代的苯基或者取代或未取代的烷基。b和b’各自独立地是取代或未取代的芳基、或者取代或未取代的杂芳基。各取代的芳基或取代的杂芳基可以被具有大于-0.50的哈米特σp值的基团取代。电子给体基团的相对强度通常由哈米特σ值、或σp值描述。用于各种取代基的哈米特σp值的列表可以在以下中找到:“asurveyofhammettsubstituentconstantsandresonanceandfieldparameters[哈米特取代基常数以及共振和场参数的测定]”,c.hansch,a.leo,以及r.w.taft,chem.rev.[化学评论],1991,91,165-195,其披露内容通过援引并入。具有大于-0.50的哈米特σp值的合适取代基的非限制性实例包括卤素基团(即,氟或溴)、烷基、全卤代烷基、苯基、甲基、苯基醚、芳烷基、乙氧基、甲氧基、对氨基苯基、芳硫基、烷硫基、酰胺、羧酸酯、芳基、杂芳基、羟基、氰基、或酯。
r1可以选自其的基团的实例包括但不限于取代或未取代的烷氧基。r2可以选自其的基团的实例包括取代或未取代的烷氧基、取代或未取代的芳氧基、取代或未取代的烷硫基、取代或未取代的芳硫基、取代或未取代的醚、取代或未取代的硫醚、氨基、或含氮杂环。b和b’可以各自独立地是取代的芳基或者取代的杂芳基。各取代的芳基或取代的杂芳基可以被具有-0.5至0.8的哈米特σp值的基团取代。b和b’可以各自独立地是取代或未取代的苯基。各苯基取代基可以选自烷氧基、卤素、烷基、或芳氧基。r1和r2合在一起可以形成环状结构,诸如环结构。
附加地或替代性地,本发明的制品可包含由式(ia)的核心骨架结构表示的吲哚萘并吡喃化合物:
关于式(ia),r1、r2、r4、b和b’如先前关于式(i)所描述的。
如以上所描述的,在没有具体示出取代基的情况下,式(ia)的核心骨架结构的其余的经编号的环位置(例如,1、2、5、和/或8)可以具有与其共价键合的氢或除氢以外的基团,例如,诸如本文所描述的基团。
进一步关于式(ia),m是0至4,并且对于各m,r3独立地是羟基;氰基;(甲基)丙烯酸酯;氨基或含氮杂环;含介晶的基团l1;取代或未取代的烷基;取代或未取代的烯基;取代或未取代的炔基;卤素基团;全卤代基团;硼酸酯或硼酸;聚醚、聚酯、聚碳酸酯、或聚氨酯;取代或未取代的芳基;取代或未取代的杂环烷基;取代或未取代的杂芳基;取代或未取代的烷氧基或者取代或未取代的芳氧基;取代或未取代的烷硫基或者取代或未取代的芳硫基;酮、醛、酯、羧酸、羧酸酯、或酰胺;碳酸酯、氨基甲酸酯、或脲;或者硅氧烷、烷氧基硅烷、或聚硅氧烷。例如,r3可以是氰基;卤素基团;卤代烷基;全卤代烷基;取代或未取代的芳基;或者取代或未取代的杂芳基。例如,r3可以在11-位置。例如,r3可以在10-位置并且是含介晶的基团l1。
进一步关于式(ia),各含介晶的基团l1可以独立地由以下式(ii)表示,
式(ii)
-[s1]c-[q1-[s2]d]d’-[q2-[s3]e]e’-[q3-[s4]f]f’-r
q1、q2、以及q3每次出现时独立地是选自由未取代的芳基、取代的芳基、未取代的环烷基、以及取代的环烷基组成的组的二价基团。芳基取代基和环烷基取代基可以各自独立地选自由以下项组成的组:液晶介晶、卤素、烷基、烷氧基、烷基氨基、二烷基氨基、烷硫基、烷氧基羰基、烷基羰基、烷氧基羰基氧基、芳氧基羰基氧基、全氟烷基、以及全氟烷氧基。进一步关于式(ii),c、d、e、以及f各自独立地是0至3的整数;并且各s1、s2、s3、以及s4每次出现时独立地选自间隔单元,所述间隔单元选自由以下项组成的组:(i)-c(z)2-、-n(z)-、-c(z)=c(z)-、-c(z)=n-,其中z每次出现时独立地选自由氢、烷基、或芳基组成的组;(ii)-si(ch3)2-、-si(ch3)2o-;以及(iii)-o-、-c(=o)-、-c≡c-、-n=n-、-s-、-s(=o)-、-(o=)s(=o)-、-(o=)s(=o)o-、-o(o=)s(=o)o-,前提是当包含杂原子的两个间隔单元连接在一起时,连接间隔单元以使得杂原子不直接彼此连接。进一步关于式(ii),r是烷基。进一步关于式(ii),d’、e’和f’各自独立地是0、1、2、3、以及4,前提是d’+e’+f’的总和至少是1。
如本文所使用的,术语“聚硅氧烷”,例如关于本发明的光致变色化合物的各个基团的取代基,包括由下式(g)表示的材料:
关于式(g),在每种情况下,下标t’是从2至200,诸如从2至100、或2至50、或从2至25、或从2至15、或从2至10、或从2至5,包括所述值。进一步关于以下式(g):对于各t’,r32和r33各自独立地选自烷基或芳基;并且r34选自氢、烷基、或芳基。在一些实施例中:对于各t’,r32和r33各自独立地选自甲基、乙基、或苯基;并且r34选自氢、甲基、乙基、或苯基。
如本文所使用的,术语“聚硅氧烷”(诸如关于本发明的光致变色化合物的各种基团的取代基、可替代地或除了由式(g)表示的材料之外)包括由以下式(h)表示的材料:
关于式(h),下标u’是0-2并且下标x’是1-3,前提是u’+x’是3;并且下标v’是0-2并且下标w’是1-3,前提是v’+w’是3。进一步关于式(h),在各情况下,对于各u’,r35独立地、对于各v’和各x’,r36独立地、以及对于各w’和各x’,各r37独立地选自烷基(诸如但不限于甲基或乙基)或芳基(诸如但不限于苯基)。
在一些实施例中,本发明的制品可包含前述式(i)和/或(ia)的化合物,单独地或与一种或多种其他光致变色化合物组合。例如,本发明的制品可包含前述式(i)和/或(ia)的化合物连同一种或多种其他光致变色化合物,这些光致变色化合物具有在300至1,000纳米范围内的活化吸收最大值。此外,上述式(i)和/或(ia)的化合物可以与一种或多种互补的常规可聚合或相容的光致变色化合物结合使用,例如像美国专利号6,113,814(在第2栏,第39行至第8栏,第41行)和6,555,028(在第2栏,第65行至第12栏,第56行)中披露的那些。
此外,式(i)和/或(ia)的化合物可以与其他光致变色化合物的混合物组合使用。例如,虽然本文没有限制,但可以使用光致变色化合物的混合物来获得某些活化的颜色。
在本发明制品的制备中可与式(i)和/或(ia)的化合物组合使用的其他光致变色化合物类别的实例包括但不限于茚并稠合萘并吡喃、萘并[1,2-b]吡喃、萘并[2,1-b]吡喃、螺芴[1,2-b]吡喃、菲并吡喃、喹啉并吡喃、荧蒽并吡喃、螺吡喃、苯并噁嗪、萘并噁嗪、螺(二氢吲哚)萘并噁嗪、螺(二氢吲哚)吡啶并苯并噁嗪、螺(二氢吲哚)荧蒽并噁嗪、螺(二氢吲哚)喹啉并噁嗪、俘精酸酐、俘精酰亚胺、二芳基乙烯、二芳基烷基乙烯、二芳基烯基乙烯、热可逆光致变色化合物和非热可逆光致变色化合物、及其混合物。可与式(i)和/或(ia)的化合物组合使用的其他光致变色化合物的其他实例可包括但不限于在us9,028,728b2的第34栏,第20行至第35栏,第13行中披露的那些。
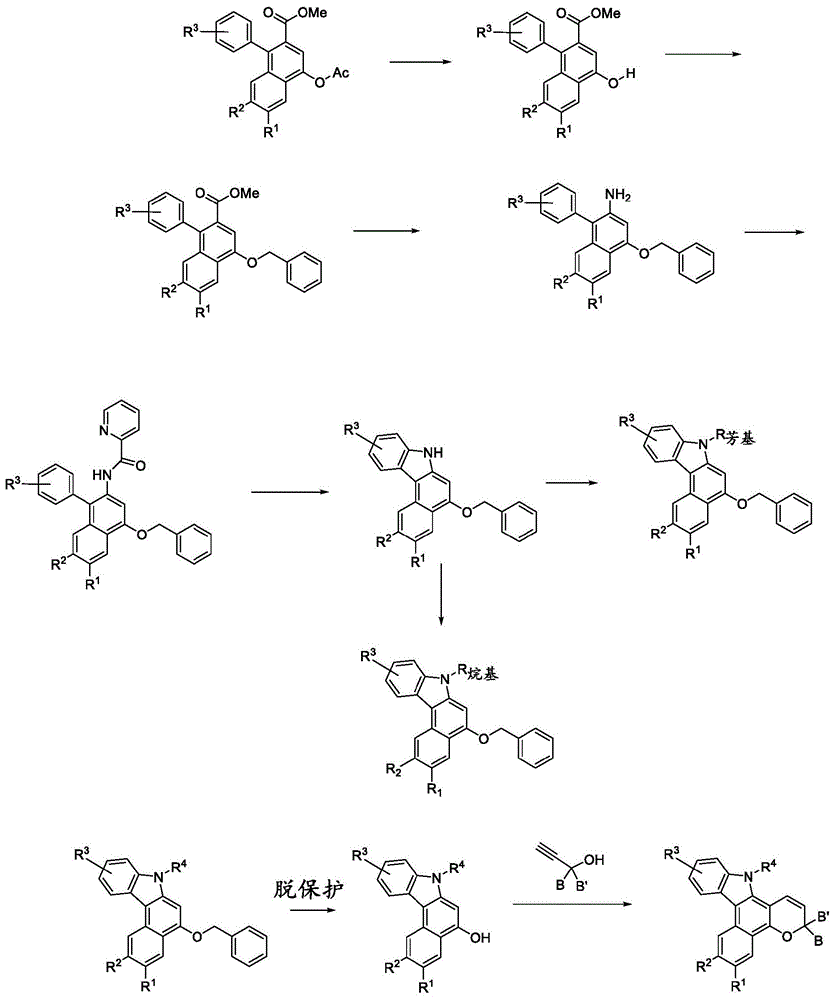
可用于本发明制品的吲哚萘并吡喃化合物可根据本领域公认的方法制备如下。出于非限制性说明的目的并参照图1、通用合成方案1,根据本发明的光致变色化合物的制备描述如下。本发明的光致变色化合物的制备的进一步详细描述在本文的实例中进一步提供。在图1中,所描绘的各种中间体、反应物、和/或化合物的各种基团,诸如r1、r2、r3、r4、b、b’、r芳基、以及r烷基各自如本文所描述的、和/或表示此类基团的前体。
在许多参考文献诸如us6,296,785或us7,262,295中已经描述了如以下式iii所描绘的具有不同取代基的化合物的合成。
羟基和羧酸基团可以通过与苄基氯和碱诸如碳酸钠或碳酸钾反应而苄基化。然后可以通过用于酯水解的酸或碱方法将所形成的羧酸酯转化成羧酸。所得产物如以下式iiia所描绘。
然后可以使用二苯基磷酰基叠氮化物经由柯提斯(curtius)重排条件将羧酸基团转化成nh2基团,该二苯基磷酰基叠氮化物产生异氰酸酯基团,随后水解以产生胺基团,如在以下式iiib中所描绘的。
通过首先通过传统的酰胺形成反应(诸如使胺与酰基氯、酯、或羧酸基团反应)形成吡啶甲酰胺基团,将胺基团转化成吲哚。胺与吡啶甲酰氯与碱诸如三乙胺的反应以高收率给出了吡啶甲酰胺,如在式iiic中所描绘的。吡啶甲酰胺可以通过使用如在以下中所描述的铜催化剂环化为吲哚,如式iiid所描绘的:takumatso,k.等人org.lett.[有机快报]2014,16,2892。参见以下所描绘的反应。
式iiid中所描绘的吲哚环也可以通过使式iiib的胺与甲苯磺酰(ts)氯或酸酐反应以形成n-ts基团而形成,如式iiie中所描绘的。该基团可以用如在以下中所描述的钯催化剂环化:youn,s.w.org.lett.[有机快报]2011,13,3738。参见以下所描绘的反应。
可替代地,式iiib的氨基可以通过在桑德迈尔(sandmeyer)条件下形成重氮盐、随后用叠氮化物的盐诸如叠氮化钠置换而转化成叠氮化物基团,如式iiif中所描绘的。然后式iiid的吲哚基团可以通过在溶剂诸如thf中暴露于uv光而形成。参见以下所描绘的反应。
如在式iiig’中所描绘的吲哚基团的烷基化可以通过在碱诸如叔丁醇钠或叔丁醇钾的存在下与烷基卤化物、三氟甲磺酸酯、或甲苯磺酸酯反应来完成。可替代地,吲哚可以由强碱诸如氢化钠或正丁基锂去质子化,并且然后使阴离子与烷基烷基卤化物、三氟甲磺酸酯、或甲苯磺酸酯反应。参见以下所描绘的反应。
通过与过渡金属催化剂和芳基卤化物的交叉偶联反应,吲哚基团可以被芳基化,如式iiig”所描绘的。用铜催化剂的乌尔曼(ullmann)偶联方法学是进行该转化的常用方法。参见以下所描绘的反应。
吲哚也可以如式iiig”所示,通过在合适的溶剂(如四氢呋喃或二甲基甲酰胺)中与芳基氟化物反应,通过亲核芳香取代进行芳基化。
苄基保护基团可以通过钯氢化条件或用强酸除去。参见以下所描绘的反应,其中式iiig是指被如本文所描述的任何r4取代的吲哚,并且经脱保护的产物示出于式iiih中。
然后式iiih中所描绘的吲哚稠合萘酚可以与芳基炔丙醇在酸性条件下反应以产出吲哚稠合萘并吡喃,如式ia中所描绘的。参见以下所描绘的反应。
如前所述并在以下实例中说明,可用于本发明制品的化合物展现出大于3:1并且在一些情况下大于6:1,如3至7:1,如3至6.5:1,如3至6.4:1,或者如3.45至6.4:1的a带与b带吸收比。
本发明的制品可以是包含一种或多种由式(i)或(ia)表示的吲哚萘并吡喃化合物的光致变色制品。光致变色制品可以通过本领域公知的方法制备,诸如通过渗吸方法、现场浇铸方法、涂覆方法、模内涂覆方法、包覆模制方法、以及层压方法。
例如,光致变色制品可以选自眼科制品、显示制品、窗、镜、有源液晶盒制品、以及无源液晶盒制品。
例如,本发明的光致变色制品可以是眼科制品,并且眼科制品可以选自矫正镜片、非矫正镜片、接触镜片、眼内镜片、放大镜片、保护性镜片、以及护目镜。
例如,本发明的光致变色制品可以是显示制品,并且显示制品可以选自屏幕、监视器、以及安全元件。
此类光致变色制品,例如光致变色镜片,在暴露于光化辐射时可以从第一未活化状态(例如,无色和非蓝色阻挡状态)转变为第二活化状态(例如,着色和蓝色阻挡状态)。在移除光化辐射源后,制品可以恢复到第一未活化(和透明或无色)状态。因此,根据本发明的光致变色制品在室外活动期间提供对与蓝光暴露相关的健康风险的增强的保护,同时在室内维持可接受的美观性。
在以下实例中更具体地描述了本发明,这些实例仅旨在作为说明性的,因为其中的许多修改和变化对于本领域技术人员将是显而易见的。
实例
下面参照以下实例1至11以及实例1a和2a描述了用于制备本发明制品的吲哚萘并吡喃化合物的制备。除非另有说明,否则化合物的结构均通过质谱法确认。
部分1:化合物的合成
实例1
步骤1
将4-乙酰氧基-6,7-二甲氧基-1-(4-(三氟甲基)苯基)-2-萘甲酸甲酯(50g,131mmol)在搅拌下分散于甲醇(200ml)中。添加浓盐酸(8ml)并将反应混合物加热至回流4小时。一旦冷却,将反应混合物浓缩至原始体积的一半,并经静置将产物沉淀。收集产物并在真空下干燥以给出41g(90%收率)的无色粉末。
步骤2
在氮气下搅拌的同时,将来自步骤1的产物(41g,101mmol)溶于无水二甲基甲酰胺(200ml)中并添加碳酸钾(28.0g,202mmol),随后添加苄基氯(15.3g,121mmol)。将反应混合物加热至70℃持续5小时,并在室温下搅拌过夜。开始形成沉淀物,并在搅拌下将反应混合物倾倒入冰水(1.0l)中。收集沉淀物并在真空下干燥以给出49.44g(99%收率)的无色固体。
步骤3
在搅拌下将来自步骤2的产物(49.4g,99.5mmol)悬浮于2-丙醇(150ml)中。添加氢氧化钠溶液(在水中10%w/w,150ml),并将反应混合物加热至回流16小时。一旦冷却,将反应混合物倾倒入冰水(1.0l)中以形成无色沉淀物。收集粉末并在真空下干燥以给出46.71g(97%收率)。
步骤4
在氮气下搅拌的同时,将来自步骤3的产物(46.7g,96.8mmol)悬浮于无水甲苯(300ml)中。添加三乙胺(25.5g,252mmol)和无水乙醇(25ml),溶解悬浮液。逐批添加叠氮化磷酸二苯酯(40g,145mmol),并将反应加热至回流3小时。一旦冷却,将反应混合物吸入200ml的乙酸乙酯中、用水(4×300ml)洗涤、用硫酸钠干燥并在减压下浓缩以给出灰白色固体(50g,98%收率)。
步骤5
将来自步骤4的产物(50.0g,95.1mmol)在搅拌下用氢氧化钠(19.2g,48.0mmol)分散在乙醇(200ml)和水(220ml)的溶液中。将反应混合物加热至回流4小时。一旦冷却,将反应混合物倾倒入冰水(1.0l)中并形成无色沉淀物。收集粉末并在真空下干燥以给出41.81g(97%收率)。
步骤6
在氮气下搅拌的同时,将来自步骤5的产物(41.81g,92.2mmol)吸入二氯甲烷(250ml)中。添加吡啶甲酸(17.0g,138.3mmol)和4-(二甲基氨基)吡啶(1.13g,9.20mmol),随后添加n,n'-二环己基碳二亚胺(22.8g,110.6mmol)。允许反应混合物在室温下搅拌4小时。过滤反应混合物并在减压下浓缩以给出微红色固体。用甲醇洗涤材料、收集并在真空下干燥以给出50.9g(99%收率)的灰白色粉末。
步骤7
在氮气下搅拌的同时,将来自步骤6的产物(50.9g,91.1mmol)溶于无水二甲基甲酰胺(100ml)中并向此添加乙酸铜(ii)(33.1g,182.2mmol)和冰乙酸(5.47g,91.1mmol)。将反应混合物加热至150℃持续20小时以给出通过高效液相色谱法确定的起始材料向产物的66%的转化率。将反应混合物经硅藻土垫过滤,并用500ml的乙酸乙酯洗涤垫。将滤液与含有乙二胺(10ml)的水(1.0l)一起添加至分液漏斗中,并分离各层。将有机层用水(3×300ml)洗涤、用硫酸钠干燥并在减压下浓缩以给出灰白色固体。使材料经受反应条件的第二次迭代和相同的分离程序。所得固体用甲醇(300ml)洗涤两次以给出灰白色粉末(37.44g,91%收率)。产物吲哚核心5-(苄氧基)-2,3-二甲氧基-9-(三氟甲基)-7h-苯并[c]咔唑通过1hnmr和质谱确认。
步骤8
在氮气下搅拌的同时,将来自步骤7的吲哚核心(5.0g,11.1mmol)溶于无水二甲基甲酰胺(40ml)中并缓慢添加氢化钠(0.8g,33.2mmol)。15分钟之后,添加碘丁烷(2.25g,12.2mmol)并将反应混合物在室温下搅拌30分钟。将反应混合物吸收在乙酸乙酯(200ml)中并用水(3×200ml)洗涤。将有机层用硫酸钠干燥并在减压下浓缩至硅胶上。进行色谱法(硅胶,在己烷中的0%-50%二氯甲烷)得到呈无色固体的产物(3.4g,60%收率)。
步骤9
在氮气下搅拌的同时,将来自步骤8的产物(1.50g,3.0mmol)与甲酸铵(0.75g,11.84mmol)和钯碳(aldrich#768243,负载在湿载体上5%,degussa型e1003u/w)在二甲基甲酰胺(20ml)中组合。将反应混合物加热至85℃持续2小时。一旦冷却,将反应混合物经硅藻土的垫过滤并用乙酸乙酯(250ml)洗涤该垫。将滤液用水(3×300ml)洗涤、用硫酸钠干燥并在减压下浓缩以给出灰白色固体,将其不经进一步纯化而使用。
步骤10
在氮气下搅拌的同时,将来自步骤9的产物(0.62g,1.48mmol)与1,1-二苯基丙-2-炔-1-醇(0.37g,1.78mmol)在甲苯(25ml)中组合并加热至75℃,添加对甲苯磺酸(5-10mg)并将反应混合物加热至回流2小时。一旦冷却,将反应混合物吸收在乙酸乙酯(25ml)中、用饱和碳酸氢钠溶液(25ml)和水(2×50ml)洗涤。将有机层用硫酸钠干燥并在减压下浓缩以给出深色油。将产物从甲基叔丁基醚、四氢呋喃和甲醇中重结晶两次,得到实例1的淡黄色粉末(0.48g,54%收率)并通过质谱法确认。
实例2
步骤1
在氮气下搅拌的同时,将实例1步骤7中所制备的吲哚核心(3.0g,6.64mmol)与溴苯(4.2g,26.6mmol)、碘化亚铜(0.63g,3.32mmol)、碳酸钾(1.82g,13.2mmol)、1,10-菲咯啉(0.24g,1.30mmol)和二苯并-18-冠-6-醚(0.24g,0.70mmol)合并在无水二甲基甲酰胺(30ml)中。将反应混合物加热至150℃持续5小时。一旦冷却,将反应混合物吸收在乙酸乙酯(250ml)中并最初用水(200ml)、用乙二胺(10ml)洗涤,随后用水(2×250ml)洗涤。将有机层用硫酸钠干燥并在减压下浓缩以给出棕色固体。将产物从甲基叔丁基醚、四氢呋喃和甲醇中重结晶两次以给出灰白色粉末(3.20g,91%收率)。
步骤2
将来自步骤1的产物(1.20g,2.27mmol)根据实例1步骤9的条件处理以给出灰白色固体,将其不经进一步纯化而使用。
步骤3
将来自步骤2的产物在实例1,步骤10的条件下处理,得到黄色粉末(1.12g,79%收率),其通过质谱法确认。
实例3-6
根据实例2制备另外的光致变色染料并总结在表1中。对于各实例,使用适当的取代的溴苯代替表1中n-偶联组分列中所指示的实例2步骤1中的溴苯。同样地,在表1中炔丙醇列中所指示的实例2步骤3中使用适当的取代的1,1-二苯基丙-2-炔-1-醇(“炔丙醇”)。通过质谱法确认结构。
实例7
实例7以与实例1相同的方式制备,不同之处在于实例1的4-乙酰氧基-6,7-二甲氧基-1-(4-(三氟甲基)苯基)-2-萘甲酸甲酯,步骤1用实例1的4-乙酰氧基-6,7-二甲氧基-1-苯基-2-萘甲酸甲酯和1,1-二苯基丙-2-炔-1-醇代替,步骤10用1-(4-甲氧基苯基)-1-苯基丙2-炔-1-醇代替。总结在表1中,并且不同的核心在吲哚核心的栏中显示。
实例8
步骤1
除了由4-乙酰氧基-7-((2,6-二甲基苯基)硫代)-6-甲氧基-1-苯基-2-萘甲酸甲酯代替4-乙酰氧基-6,7-二甲氧基-1-(4-(三氟甲基)苯基)-2-萘甲酸甲酯之外,根据实例1和2的条件制备起始中间体,并总结于表1中。
步骤2
在氮气下搅拌的同时,将步骤1的产物(1.46g,2.58mmol)与对甲苯磺酸(0.45g,2.58mmol)合并在甲苯(30ml)中并加热至85℃持续1小时。一旦冷却,将反应混合物用水(2×75ml)洗涤、用硫酸钠干燥并在减压下浓缩以给出黄色固体(产物与副产物的1:2混合物)。将固体悬浮在甲苯(20ml)中并添加1,1-二苯基丙-2-炔-1-醇(0.36g,1.73mmol)。将反应混合物加热至回流并添加对甲苯磺酸(5mg-10mg)。在回流下将反应混合物加热1小时后,使反应混合物冷却并在减压下浓缩到硅胶上。进行色谱法(硅胶,在己烷中的0%-50%二氯甲烷)得到黄色固体。从甲基叔丁基醚、四氢呋喃和甲醇中重结晶给出浅黄色粉末(0.23g,44%收率,基于混合物),将其通过质谱法确认。
实例9
根据实例1和2的条件制备实例9,除了将4-乙酰氧基-6,7-二甲氧基-1-(4-(三氟甲基)苯基)-2-萘甲酸甲酯替换为4-乙酰氧基-6-甲氧基-1-苯基-2-萘甲酸甲酯,并总结在表1中。
实例10
根据实例1的条件制备实例10,除了将4-乙酰氧基-6,7-二甲氧基-1-(4-(三氟甲基)苯基)-2-萘甲酸甲酯替换为4-乙酰氧基-6-甲基-1-(对甲苯基)-2-萘甲酸甲酯,并总结在表1中。
实例11
根据实例1的条件制备实例11,除了将4-乙酰氧基-6,7-二甲氧基-1-(4-(三氟甲基)苯基)-2-萘甲酸甲酯替换为4-乙酰氧基-1-苯基-2-萘甲酸甲酯,并总结在表1中。
如下所述制备实例1a和2a并总结在下表2中。除非另有说明,否则所得产物结构均通过质谱法确认。
实例1a
步骤1
在氮气下搅拌的同时,将实例2,步骤2的产物(1.27g,2.90mmol)与1-(4-丁氧基苯酚)-1-苯基丙-2-炔-1-醇(0.37g,1.78mmol)在甲苯(25ml)中组合并加热至75℃,添加对甲苯磺酸(5-10mg)并将反应混合物加热至回流3小时。一旦冷却,将反应混合物吸收在乙酸乙酯(200ml)中,用饱和碳酸氢钠溶液(100ml)和水(2x100ml)洗涤。将有机层用硫酸钠干燥并在减压下浓缩至硅胶上。进行色谱法(硅胶,己烷中的0-70%乙酸乙酯)得到灰白色粉末。将产物从甲基叔丁基醚、四氢呋喃和甲醇中重结晶两次,得到无色粉末(1.21g,60%收率)并通过1hnmr光谱法和质谱法确认。
步骤2
在氮气下搅拌的同时,将二甲基哌啶(0.33g,2.90mmol)添加到无水四氢呋喃(35ml)中。缓慢添加正丁基锂(在己烷中2.5m,1.2ml)并使混合物在室温下搅拌5分钟。添加步骤1的产物(0.5g,0.71mmol)并使反应混合物搅拌附加的4小时。将反应混合物倾倒入水(250ml)中并用乙酸乙酯(2×50ml)萃取。将有机层用硫酸钠干燥并浓缩至硅胶上。进行色谱法(硅胶,己烷中的0至50%二氯甲烷)得到呈灰白色固体的产物(0.096g,20%收率),其通过质谱法确认。
实例2a
步骤1
将来自实例7的吲哚核心,5-(苄氧基)-2,3-二甲氧基-7h-苯并[c]咔唑用1-溴-2-乙基己烷代替碘丁烷处理至实例1,步骤8的条件。
步骤2
在氮气下搅拌的同时,通过注射器将甲基格氏(grignard)溶液(1.4m,29.0ml)缓慢添加至二甲基哌啶(4.93g,40.0mmol)中,并允许反应混合物搅拌10分钟。将来自步骤1的产物(4.95g,10.0mmol)溶解在无水四氢呋喃(30ml)中,并经10分钟逐滴添加至格氏(grignard)/二甲基哌啶混合物中。一旦添加,将反应混合物回流3小时。一经冷却,将反应混合物倾倒入1mhcl溶液(75ml)中,并将水层用乙酸乙酯(3×150ml)萃取。将有机层合并、用硫酸钠干燥并在减压下浓缩至二氧化硅上。进行色谱法(硅胶,己烷中的0-100%二氯甲烷)得到半固体(2.17g,44%收率)。
步骤3
将步骤2的产物(2.17g,4.50mmol)根据实例1步骤9的条件处理以给出泡沫,将其不经进一步纯化而使用。
步骤4
在氮气下搅拌的同时,将步骤3的产物与1,1-双(4-氟苯基)丙-2-炔-1-醇合并,并溶解在甲苯(40ml)中。将混合物加热至回流并添加对甲苯磺酸(10mg)。将反应混合物在回流下加热5小时直至通过高效液相色谱法确定反应的转化率是60%,并且一旦冷却,浓缩至硅胶上。进行色谱法(硅胶,在己烷中的5%-50%乙酸乙酯)得到棕色/绿色玻璃状物(0.85g,30%收率)。
步骤5
在氮气下搅拌的同时,将步骤4的产物(0.85g,1.38mmol)溶解于无水二氯甲烷(25ml)中。添加无水吡啶(0.5ml,5.52mmol),并将反应混合物在冰浴中冷却。逐滴添加三氟甲磺酸酐(0.51g,1.80mmol)。30分钟之后,将反应混合物倾倒入水中并分离各层。将水层用二氯甲烷(2×30ml)洗涤。将有机层合并、用硫酸钠干燥并在减压下浓缩以给出棕色/绿色玻璃状物(1.0g,97%收率)。
步骤6
将来自步骤5的产物(1.0g,1.33mmol)与苯甲酰胺(0.25g,2.0mmol)、碳酸铯(1.80g,5.52mmol)和4,5-双(二苯基膦基)-9,9-二甲基呫吨(xantphos)合并在甲苯和乙醇溶液(30ml,9:1v/v)中,并在氮气下鼓泡10分钟。添加三(二亚苄基丙酮)二钯(0)(0.12g,0.13mmol),并将反应混合物加热至回流2小时。将反应混合物吸收在乙酸乙酯(100ml)中、用水(2×100ml)洗涤、用硫酸钠干燥并浓缩至硅胶上。进行法(硅胶,在己烷中的0%-30%乙酸乙酯)得到棕色固体。从甲醇中重结晶得到0.38g(40%收率)的浅黄色粉末。
部分2:测试
如美国专利号8,608,988实例1-3所述,将来自实例1至11以及1a和2a,以及每个对比实例ce1至ce4的光致变色染料中的每一种以相同的mol%掺入聚氨酯涂料体系中,并以相同的涂层厚度施加到由
通过首先在约14厘米的距离处暴露于365纳米紫外光10分钟以活化涂层内的光致变色材料来调节每个经涂覆的测试芯片(以下称为“测试样品”)。用
根据cie15:2004比色法,使用d65光源和10°观测器,使用ciey值确定实例1至11、1a和2a以及每个对比实例ce1至ce4的透射率百分比。如本文在说明书和权利要求书中所使用的a*和b*值是指根据cie15:2004空间色度法,采用d65光源和10°观测器,使用hunterultrascanpro装置测量的a*和b*值。根据iso12311:2013(e),第7.4小节确定光谱蓝光透射率百分比(在本文中报告为τsb)。
bmp光学平台配备有两个彼此成直角的150-瓦
根据光密度从未活化或发白状态到活化或着色状态的变化(δod),通过建立初始未活化透射、打开来自一个或多个氙灯的遮光器并在选定的时间间隔测量通过活化的透射来确定响应测量。根据下式确定光密度变化:δod=log(10)(%tb/%ta),其中%tb是在发白状态下的百分比透射,%ta是在活化状态下的百分比透射。饱和下δod是在活化15分钟之后,并且褪色半衰期(“t1/2”)值是在移除活化光源之后涂层中的光致变色材料的活化形式的δod在73.4°f(23℃)下达到十五分钟δod的一半的时间间隔,以秒为单位。
疲劳测试:
在初始性能测试之前,用于疲劳测试的程序和结果在光学平台上确定。为了活化光致变色分子,通过将测试样品在距离源约14cm处暴露于365nm紫外光下15分钟来调节测试样品。用licor型号li-1800分光辐射计测量测试片处的uva辐照度,并且发现是22.2瓦/平方米。然后将测试样品置于75℃的烘箱中1小时。然后将测试样品暴露于室内灯下3小时。最后,然后将测试样品在测试前保持在黑暗环境中至少1小时,以便在测试前继续褪色回到基态。
将配备有schott3mmkg-2带通滤光片、一个或多个中性密度滤光片和newportmodel#67005300-瓦特氙弧灯(具有model#69911电源和newportmodel689456数字曝光/定时器)的光学平台用于控制用于活化每个涂覆的测试样品的辐照光束的强度。uniblitzmodel#cs25s3zmo高速快门,其带有型号#vmm-d3控制器,以及用于活化光束准直和聚焦的熔融石英聚光透镜,通过石英水囊/样品架来保持样品温度,每个待测试的测试样品都插入其中。用泵水循环系统来控制水池中的温度,其中水穿过放置在制冷单元的贮存器中的铜盘管。用于保持测试样品的水囊在正面和背面都包含熔融石英片,以消除活化或监测光束的光谱变化。将穿过水池的经过滤的水维持在100°f±2°用于在暴露于atlas老化试验仪之前和之后的光致变色测试。
将用于监测响应测量的定制宽带光源以垂直于池组件的表面的方式定位。该宽束光源通过收集和合并来自100-瓦卤钨灯(由lambdaup60-14恒压电源控制)的单独过滤的光与分裂端、分叉的纤维光缆以增强短波长光强度来获得。在穿过测试样品之后,将该监测光重新聚焦至2-英寸积分球中并由纤维光缆馈送至海洋光学公司(oceanoptics)s2000分光光度计。oceanopticsspectrasuite和专有软件用于测量响应和控制光学平台的操作。
使用具有包含型号sed033检测器、b滤光器和漫射器的检测器系统的国际光研究辐射计、型号il-1700以验证测试之前的辐照度。将18.0w/m2的调整值用作辐照度验证设置点。相对于licor1800-02光学校准校准器校正辐射计的输出显示(因子值设置),以便显示表示瓦特每平方米uva的值。增加或减少经过控制器至灯的电流和/或通过在活化光路径中添加或移除中性密度滤光器来对氙气灯输出进行调整。将测试样品以与测试样品表面垂直的方向呈31°暴露于活化光下。
从脱色的第一状态到着色的第二状态的光密度变化(δod)是通过建立初始透射率来确定的,打开氙灯的快门以提供紫外线辐射,使测试样品从脱色的第一状态变为活化的第二(即,着色)状态,并在典型地活化5分钟后测量活化状态下的透射率。使用以下公式计算光密度的变化:δodlog(%tb/%ta),其中%tb是在发白的第一状态下的百分比透射,%ta是在活化状态下的百分比透射,并且对数是以10为底。这提供了od初始。
使用atlasci4000老化试验仪用于进行模拟太阳辐射加速老化。测试样品被暴露在1小时的黑暗循环以及然后使用硼/硼硅酸盐过滤的氙弧灯(在340nm下每平方米输出0.25瓦)的65小时光循环。将老化试验仪中的温度维持在45℃下,并且将相对湿度控制在70%湿度下。将黑色板的温度维持在55℃下。
在测试样品经历该uv暴露疲劳循环后,将这些样品如上所述进行预处理并在光学平台上进行测量,以在与初始测试所述相同的条件下获得最终δod最终。
疲劳百分比是根据下式,通过测量加速风化前后测试样品的光密度变化(δod)之间的差异来确定的:%疲劳=(δod初始-δod最终/δod初始)×100。
饱和下δod是在活化15分钟之后,并且褪色半衰期(“t1/2”)值是在移除活化光源之后涂层中的光致变色-二向色性材料的活化形式的δod在73.4°f(23℃)下达到十五分钟δod的一半的时间间隔,以秒为单位。
在完全活化下,观察到对于本发明的吲哚萘并吡喃化合物的两个吸收最大值δod。波长a(“λa”)是具有在420nm-500nm之间的最大δod的波长,并且波长b(“λb”)是具有在500nm-650nm之间的最大δod的波长。a带与b带吸收比(“a-b比”)由下式计算:
a-b比率=最大δod波长a/最大δod波长a
测试结果在下表3中呈现。
上表3中呈现的数据说明,用于制备本发明制品的光致变色吲哚萘并吡喃化合物提供了在第二状态下提供优异的(低)蓝光透射率(报告为τsb)的制品,同时与在活化(第二)状态下具有类似黄色的对比实例ce1至ce4的本领域公认的光致变色化合物相比,保持了非常好的动力学和疲劳特性
本发明可以进一步地由以下非限制性条款中的一项或多项表征。
条款1.一种制品,其响应于电磁辐射从第一状态转变为第二状态,该制品包含吲哚萘并吡喃,其中,
在该第一状态下,该制品展现出大于80%的透射率百分比,以及-5至15,如-5至10,如-5至5的b*值;并且
在该第二状态下,该制品展现出在35%与75%之间的透射率百分比,以及小于20%的τsb值。
条款2.如条款1所述的制品,其中,在该第一状态下,该制品展现出大于80%的透射率百分比,以及0至15,如0至10,如0至5的b*值。
条款3.如条款1或2所述的制品,其中,在该第二状态下,该制品展现出在35%与75%之间的透射率百分比,以及3.0%至小于20%,如4.0%至小于20%,如5.0%至小于20%的τsb值。
条款4.如条款1至3所述的制品,其中,该吲哚萘并吡喃是光致变色吲哚萘并吡喃,其包含由下式(i)表示的核心骨架结构:
其中,
r1和r2各自独立地是氢、取代或未取代的烷氧基、取代或未取代的芳氧基、取代或未取代的烷硫基、取代或未取代的芳硫基、取代或未取代的醚、取代或未取代的硫醚、氨基、含氮杂环、取代或未取代的烷基、取代或未取代的芳基、-nhc(o)ra、或-oc(o)ra,
其中ra是取代或未取代的烷基、取代或未取代的芳基、取代或未取代的杂芳基、取代或未取代的烷氧基、取代或未取代的芳氧基、取代或未取代的烷硫基、或者取代或未取代的芳硫基;
r4选自氢、取代或未取代的烷基、取代或未取代的杂环烷基、烯丙基、取代或未取代的芳基、或者取代或未取代的杂芳基;并且
b和b’各自独立地是取代或未取代的芳基、或者取代或未取代的杂芳基,其中各取代的芳基或取代的杂芳基被具有大于-0.50的哈米特σp值的基团取代。
- 还没有人留言评论。精彩留言会获得点赞!