近平滑假丝酵母酸性蛋白酶SAPP1及表达、纯化方法与流程
本发明属于生物工程技术领域,特别涉及近平滑假丝酵母酸性蛋白酶sapp1及表达、纯化方法。
背景技术:
假丝酵母菌是世界上8%~10%医院获得性血流感染的致病因子,白假丝酵母是最常见的,但其他物种(如近平滑假丝酵母candidaparapsilosis)的感染频率也在增加。近平滑假丝酵母是拉丁美洲第二最常见的假丝酵母,在1岁以下的儿童中尤其流行。近平滑假丝酵母也是欧洲常见的感染源。近平滑假丝酵母的发病机制与使用留置医疗器械和高糖饲料有关,已经成为败血症和免疫受损患者伤口和组织感染的一个重要原因。这一物种经常被发现存在于医护人员手中,并且已经导致了几起暴发感染。
与白色假丝酵母菌和热带假丝酵母菌不同,近平滑假丝酵母不是一种专性的人类病原体,它是从非人类来源如家畜、昆虫或土壤中分离出来的。近平滑假丝酵母也是一种正常的人类共生菌,是最常分离的真菌之一。免疫功能低下的个体和外科手术患者,尤其是那些胃肠道手术的患者,感染近平滑假丝酵母的风险很高。目前对于治疗由c.parapsilosis引起的侵袭性念珠菌病尚无共识,对其感染机制也没有非常清晰的研究,通常认为其分泌的酸性蛋白酶在破坏宿主角质层、水解基质和蛋白质中起重要作用。目前的研究中主要集中在白色假丝酵母上,对近平滑假丝酵母的研究极少,重组表达并纯化假丝酵母分泌酸性蛋白酶几乎没有,其酸性蛋白酶的表达纯化对研究假丝酵母感染机制及寻找潜在治疗方法具有重要意义。
另一方面,近平滑假丝酵母也是一种重要的工程菌。魏文婷等利用近平滑假丝酵母作为生产甘露醇的工程菌;谢鑫磊等利用近平滑假丝酵母作为生产油脂的工程菌;李亚莉等以晒青茶为原料,接种近平滑假丝酵母发酵普洱茶生产gaba;许敏伟利用近平滑假丝酵母作为立体异构生产(s)-ped的反应介质。近平滑假丝酵母来源的酸性蛋白酶在营养和致病方面都有重要功能,而且在酸性条件下具有高活性和高稳定性的蛋白酶,具有重大的工业应用前景。目前重组表达假丝酵母酸性蛋白酶的产量较低,如zuzanavinterová等在酿酒酵母里表达近平滑假丝酵母sapp1的产量为3mg/l;e.hochuli等在大肠杆菌表达sap2的酶活为2.3u/mg;jingli等在大肠杆菌表达sap6的酶活为19.6u/mg。
技术实现要素:
本发明的首要目的在于克服现有技术的缺点与不足,提供一种近平滑假丝酵母酸性蛋白酶sapp1,该酸性蛋白酶属于a1家族,发酵液蛋白酶活为176.9u/ml,比酶活为1478.3u/mg,高于同类报道的酸性蛋白酶酶活。
本发明的另一目的在于提供所述的酸性蛋白酶sapp1的异源表达及纯化方法。本发明以近平滑假丝酵母为来源,采用了基因工程的技术方法,从中获得酸性蛋白酶基因sapp1,构建重组质粒,然后在毕赤酵母中重组表达,通过亲和层析对发酵液中的酸性蛋白酶组分进行纯化,首次在毕赤酵母中成功表达纯化了近平滑假丝酵母酸性蛋白酶sapp1。
本发明的再一目的在于提供一种重组表达载体、重组工程细胞。
本发明的目的通过下述技术方案实现:
一种近平滑假丝酵母酸性蛋白酶sapp1,其氨基酸序列如seqidno.1所示。
编码所述的近平滑假丝酵母酸性蛋白酶sapp1的核苷酸序列优选如seqidno.2所示。
所述的近平滑假丝酵母酸性蛋白酶sapp1的异源表达及纯化方法,包括如下步骤:
(1)构建含有所述的酸性蛋白酶基因片段sapp1的重组表达载体;
(2)将所述的重组表达载体转化细胞;
(3)对阳性细胞进行发酵诱导及纯化。
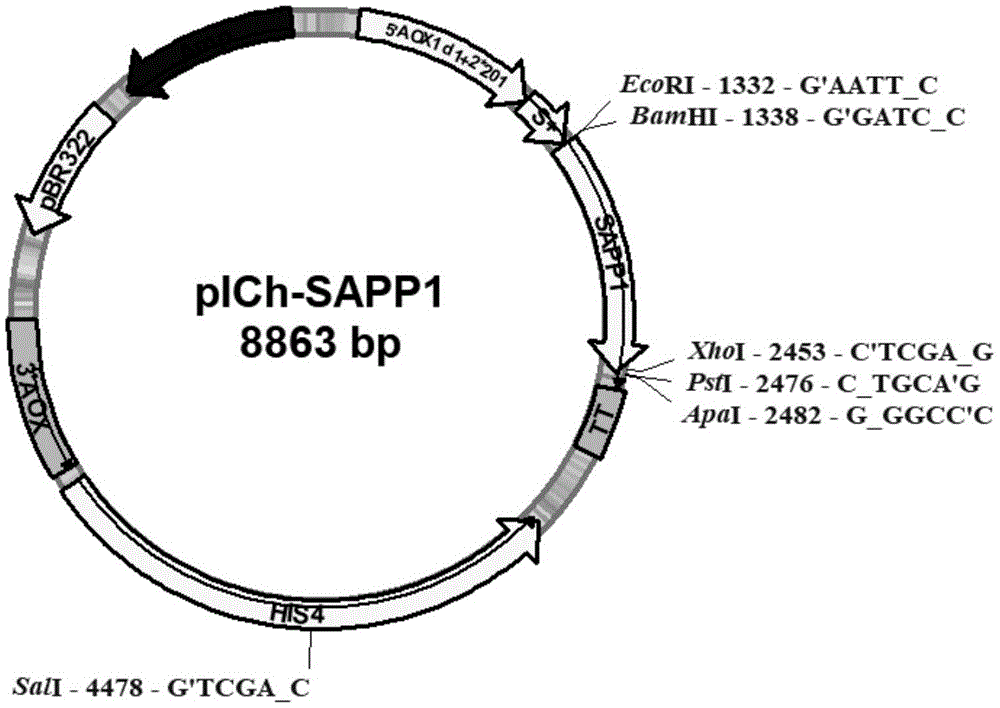
步骤(1)中所述的表达载体优选为表达载体pich,该表达载体通过将表达载体ppic9k启动子改造为dl+2x201aoxl,其核苷酸序列如seqidno.5所示;优化α-factor信号肽编码序列如seqidno.6所示,去除载体ppic9k当中3个psti和1个xhoi,突变amp抗性基因7068t>g,删除了kan抗性片段,并将多酶切位点改造替换为ecori,bamhi,mlui,xhoi,psti,apai得到。
所述的酸性蛋白酶基因片段sapp1与表达载体pich片段优选按摩尔比为1:5进行连接。
步骤(1)中所述的酸性蛋白酶基因片段sapp1优选通过提取近平滑假丝酵母总rna,反转录得到近平滑假丝酵母cdna,再以所述的近平滑假丝酵母cdna为模板,设计特异性引物通过pcr扩增得到。
所述的特异性引物优选为正向引物f(5’-gtcaggatccattccagaggaggctgctaa-3’)和反向引物r(5’-atgcctgcagaacggcagaaatgctcga-3’),下划线序列分别表示bamhi酶切位点和psti酶切位点。
所述的近平滑假丝酵母优选为近平滑假丝酵母gim2.146。
所述的近平滑假丝酵母优选培养于ypd固体培养基中,30℃培养2天。
所述的ypd固体培养基的配方优选为葡萄糖2g,蛋白胨2g,酵母提取物1g,琼脂粉1g。
步骤(2)中所述的细胞优选为毕赤酵母gs115。
步骤(2)中所述的转化优选为将所述的重组表达载体线性化后通过电转化法转入细胞。
所述的线性化的操作优选为用sali内切酶酶切所述的重组表达载体。
步骤(2)中所述的将所述的重组表达载体转化细胞的具体步骤进一步优选为:
①构建并提取pmd18-t-sapp1克隆载体质粒,以之为模板进行pcr扩增,鉴定产物后用纯化回收,得到回收产物;
②把表达载体ppic9k启动子改造为dl+2x201aoxl,其核苷酸序列如seqidno.5所示;优化α-factor信号肽编码序列如seqidno.6所示,去除载体ppic9k当中3个psti和1个xhoi,突变amp抗性基因7068t>g,删除了kan抗性片段,并将多酶切位点改造替换为ecori,bamhi,mlui,xhoi,psti,apai,构建得到表达载体pich;
③使用bamhi和psti对步骤①的回收产物和步骤②得到的pich载体进行双酶切后,分别纯化回收,得到双酶切后的蛋白酶基因片段和pich载体;
④将步骤③中得到的蛋白酶基因片段和pich载体进行体外连接,构建得到表达质粒pich-sapp1;
⑤对表达质粒pich-sapp1,用sali内切酶酶切,将表达质粒载体线性化;
⑥制备毕赤酵母gs115感受态;使用电转化法把步骤⑤得到的线性化质粒转入毕赤酵母中。
步骤①中所述的鉴定优选为通过琼脂糖凝胶电泳进行鉴定。
步骤①中所述的构建pmd18-t-sapp1克隆载体质粒的具体操作为:将酸性蛋白酶基因片段sapp1与pmd18-t载体连接,t载体连接体系如下:pmd18-t为0.5μl、sapp1为4.5μl、solutioni为5μl。
步骤④中所述的蛋白酶基因片段和pich载体优选按摩尔比为1:5配比进行体外连接。
步骤(3)中所述的纯化优选为使用镍柱亲和层析纯化目的蛋白;所述的镍柱亲和层析所用洗脱液优选为0.15m咪唑、0.5mnacl、0.02m磷酸缓冲液(ph7.4)。
步骤(3)中所述的发酵诱导的条件优选为在bmmy培养液中,28℃,250rpm振荡培养,每24小时添加1.5%的甲醇溶液进行诱导表达;诱导的时间优选为7天。
所述的bmmy培养液的配方优选为酵母提取物10g,胰蛋白胨20g,ynb13.4g,甲醇10ml,1m磷酸钾(ph6.0)100ml,蒸馏水定容至1000ml。
一种重组表达载体,含有所述的酸性蛋白酶sapp1的核苷酸序列。
一种重组工程细胞,含有所述的重组表达载体;该重组工程细胞可以表达所述的酸性蛋白酶sapp1。
本发明相对于现有技术具有如下的优点及效果:
1.本发明首次在毕赤酵母中成功表达纯化了近平滑假丝酵母酸性蛋白酶sapp1。
2.本发明得到的sapp1蛋白酶酶活远高于现有同类的酸性蛋白酶。
采用国标中的福林法测定sapp1的蛋白酶活,测得在ph5.0和40℃条件下发酵液蛋白酶活为176.9u/ml,比酶活为1478.3u/mg,高于同类报道的酸性蛋白酶酶活,如zuzanavinterová等在酿酒酵母里表达近平滑假丝酵母sapp1的产量为3mg/l;e.hochuli等在大肠杆菌表达sap2的酶活为2.3u/mg;jingli等在大肠杆菌表达sap6的酶活为19.6u/mg;且本发明的sapp1蛋白酶的水解效果优于商业木瓜蛋白酶及酸性蛋白酶。
3.本发明的方法大幅度提高了sapp1蛋白酶的产量,产量可高达312.5mg/l发酵液。
附图说明
图1是重组表达载体pich-sapp1的质粒图谱。
图2是重组蛋白的sds-page凝胶电泳图,其中,m表示标准分子量蛋白,泳道1是经过7天发酵诱导后得到的粗酶液,泳道2是经过纯化后的样品,泳道3是经过7天诱导的转化空载体的毕赤酵母上清液。
图3是水解实验结果照片图,其中1为稀释到100u/ml实施例2得到的发酵液,2为100u/ml的商业木瓜蛋白酶(奥博星),3为100u/ml的商业酸性蛋白酶(夏盛),4为灭活的稀释到100u/ml实施例2得到的发酵液。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1近平滑假丝酵母sapp1蛋白酶的异源表达
(一)近平滑假丝酵母培养
将近平滑假丝酵母gim2.146(购于广东省微生物菌种保藏中心)接种至ypd固体培养基(葡萄糖2g,蛋白胨2g,酵母提取物1g,琼脂粉1g)中,30℃培养2天。
(二)近平滑假丝酵母总rna提取
(1)用高压蒸汽灭菌后的镊子摄取约100mg的假丝酵母放入液氮预冷的研钵中,加入少量液氮,用研钵快速研磨,再加入少量液氮,继续研磨,反复3次,直至全部假丝酵母彻底变成白色粉末。
(2)向研钵中加入2mlrnaisoplus(购于大连宝生物工程有限公司),尽量将粉末完全覆盖,然后室温静置,直至rnaisoplus完全融化,用研钵继续研磨至裂解液呈透明状。将所得的裂解液等量转移至1.5ml离心管中,室温静置5分钟。12000rpm,4℃离心5分钟,小心吸取上清液,移入新的离心管中(切勿吸取沉淀)。
(3)向步骤(2)中得到的上清液加入400μl氯仿,盖紧离心管盖,剧烈振荡15秒。待溶液充分乳化后,再室温静置数分钟后12000rpm,4℃离心15分钟。
(4)从离心机中小心取出离心管,此时匀浆液分为三层,无色的上清液、中间的白色蛋白层和带有颜色的下层有机相;吸取上清液转移至另一新的离心管中。
(5)向步骤(4)得到的上清液中加入等体积的异丙醇,上下颠倒离心管充分混匀后,在室温下静置10分钟后,12000rpm,4℃离心10分钟。
(6)离心之后,试管底部有沉淀。小心弃去上清,缓慢地沿离心管壁加入1ml75%的乙醇(切勿触及沉淀),轻轻上下颠倒,洗涤离心管管壁,12000rpm,4℃离心5分钟后小心弃去乙醇。
(7)打开离心管盖子,倒置管子,室温干燥沉淀5分钟,加入20μl的rnase-free水溶解沉淀,待沉淀完全溶解后,将溶解液转移至rnase-free离心管中,置于-80℃保存。
(三)rt-pcr克隆sapp1编码序列:
(1)使用primescript1ststrandcdnasynthesiskit试剂盒(大连宝生物工程有限公司)进行反转录,把下列组分加入到一个无核酶的pcr管中:
表1rt-pcr体系
(2)65℃保温上述混合物5分钟后迅速置于冰上,再加入4μl5×first-strandbuffer,2μl0.1mdtt,1μl40u/mlrnase抑制剂于上述混合物中,轻轻混合并在37℃培养2分钟;
(3)加入1μl反转录酶,轻轻混合后在37℃培养50分钟;
(4)70℃培养15分钟灭活反转录酶,置于-20℃保存;
(5)设计正向引物f(5’-gtcaggatccattccagaggaggctgctaa-3’)和反向引物r(5’-atgcctgcagaacggcagaaatgctcga-3’)。以上一步获得的假丝酵母cdna为模板,按照以下pcr体系及程序,进行pcr反应获得目的dna片段;
表2pcr体系
pcr反应条件为:94℃预变性3min;94℃变性30秒;51℃退火30秒;72℃延伸2分钟;32个循环;最后72℃延伸10分钟;然后将产物通过琼脂糖凝胶电泳鉴定,琼脂糖浓度为1.5%,电泳条件为120v,25min,琼脂糖凝胶电泳的操作过程参见《分子克隆实验指南》。得到酸性蛋白酶基因条带大小约为1200bp,使用凝胶回收试剂盒回收目的基因。
(四)ta克隆与重组质粒的筛选鉴定
(1)高保真酶产物经切胶回收后,利用ex-taq酶进行加a反应,按以下体系将步骤(三)得到的目的基因片段(sapp1)与pmd18-t载体(购于大连宝生物工程有限公司)连接,连接条件16℃,2h;
表3t载体连接体系
然后42℃热击70秒转化大肠杆菌dh5α感受态细胞(购买于大连宝生物工程有限公司),涂布含有氨苄青霉素抗性的lb液体培养基(胰蛋白胨10g,酵母提取物5g,氯化钠10g,蒸馏水定容至1000ml)中,37℃培养过夜。然后使用2×taqpcrmix进行菌落pcr筛选阳性菌落,反应体系及程序如下:
表4菌落pcr体系
菌落pcr反应条件为:94℃预变性3min;94℃变性30秒;51℃退火30秒;72℃延伸1分钟;32个循环;最后72℃延伸10分钟;然后将产物通过琼脂糖凝胶电泳鉴定,琼脂糖浓度为1%,电泳条件为120v,25min,得到酸性蛋白酶基因条带大小约为1200bp;
(2)挑取阳性克隆加入到含氨苄抗性的lb液体培养基于37℃,220rpm摇床上扩大培养12h。取扩大培养的菌液于离心管中,送至生工生物工程(上海)股份有限公司进行测序,经过测序测得克隆基因长度为1128bp,其核苷酸序列如seqidno.2所示。
(五)基因sapp1转化毕赤酵母gs115:
(1)提取克隆有酸性蛋白酶基因的pmd18-t-sapp1克隆载体质粒,以之为模板进行pcr扩增,pcr体系参照前文表2,然后将产物进行琼脂糖凝胶电泳鉴定,用试剂盒纯化回收。把表达载体ppic9k启动子aoxl改造为dl+2x201aoxl(将野生型a0x1启动子序列的-777到-712的序列缺失,并重复序列-203到-190的序列,所述的dl+2x201aoxl的核苷酸序列如seqidno.5所示),分析毕赤酵母密码子偏好性优化α-factor信号肽编码序列为如seqidno.6所示,上述两步皆通过基因合成片段后对载体进行rf克隆改造。去除载体ppic9k当中3个psti和1个xhoi,突变amp抗性基因7068t>g(指第7068个碱基从t突变为g,使用aacgttgttgccattgcggcaggcatcgtggtgtcacg对载体进行rf克隆),使用psti酶切后重新连接删除了kan抗性片段和tacttgaagtcggacagtgagtgtagtcttgagaaattctgaagccgtatttttattatcagtgagtcagt对载体进行rf克隆,并将多酶切位点改造替换为ecori,bamhi,mlui,xhoi,psti,apai,构建得到表达载体pich。
使用bamhi和psti对回收产物和pich载体按照说明书进行双酶切(双酶切体系如表5),用普通dna回收试剂盒对酶切后的dna分别进行纯化回收,然后用核酸浓度检测仪检测dna浓度;
表5双酶切体系
酶切条件37℃,4h;
(2)将纯化回收的蛋白酶基因片段sapp1和pich载体片段按摩尔比为1:5的比例利用t4连接酶进行体外连接,连接条件22℃,8h,连接体系如下表。
表6t4连接酶连接体系
将连接后的重组质粒42℃热击70秒转化至大肠杆菌dh5α菌株中。在含有氨苄的平板上挑选阳性单菌落,提取其质粒进行双酶切鉴定,并且对该菌株也进行菌液和质粒测序鉴定;对构建成功的表达质粒命名为pich-sapp1。
(3)采用质粒小提试剂盒从阳性克隆菌株的菌悬液中提取表达质粒载体,然后对表达质粒载体用sali内切酶酶切,将表达质粒载体线性化,酶切体系如下:
表7单酶切体系
酶切之后,用回收试剂盒回收质粒并测定浓度;
(4)制备毕赤酵母gs115感受态,具体步骤如下:
a.将-80℃冻存的毕赤酵母gs115菌株(购于invitrogen)于ypd平板(胰蛋白胨20g,酵母提取物10g,葡萄糖20g,琼脂粉20g,蒸馏水定容至1000ml)上划线接种,30℃培养3天;
b.挑取毕赤酵母gs115菌株的单菌落接种至含有25mlypd培养液(胰蛋白胨20g,酵母提取物10g,葡萄糖20g,蒸馏水定容至1000ml)的250ml三角瓶中,30℃、250rpm振荡培养2天;
c.取1ml步骤b最终得到的菌悬液接种至另一瓶含有50mlypd培养液三角瓶中,30℃、250rpm振荡培养数小时,至菌悬液的od600达到2.0;
d.将菌悬液转移已灭菌的50ml离心管中,5000rpm,4℃离心5分钟,去上清,用预冷的无菌水10ml将菌体重悬,然后转移至15ml离心管中;
e.5000rpm,4℃离心5分钟,去上清,用预冷的1m山梨醇10ml将菌体重悬,重复一次;
f.5000rpm,4℃离心5分钟,去上清,用1ml1m山梨醇重悬菌体,置于冰上用于电转化;
(5)使用电转化法把步骤(3)得到的线性化质粒转入毕赤酵母中,电击条件为1.5kv,2mm电转杯,10ng线性化质粒。把电转化后的菌液涂布md平板,30℃培养2天,挑选5个单菌落分别接种ypd液体培养基,然后低温裂解毕赤酵母转化子细胞,
裂解方法如下:
a.从md平板(葡萄糖20g,ynb13.4g,琼脂粉20g,蒸馏水定容至1000ml)上,挑取10个毕赤酵母转化子单菌落分别接种至2mlypd培养液中,30℃,220rpm振荡培养2天;
b.分别将1ml菌液转移至离心管中,8000rpm离心2min,弃上清;
c.加入1mltebuffer重悬菌体,8000rpm离心2min,弃上清,重复一次;
d.沸水浴30min后转移至-80℃冰箱放置一个小时,然后沸水浴10min;
e.8000rpm离心2min,所得上清置于-20℃保存;
使用2×taqpcrmix进行菌落pcr鉴定,反应体系及程序参照表4。
实施例2蛋白酶sapp1发酵诱导、纯化与酶活测定
挑选实施例1得到的阳性重组毕赤酵母菌株划线md平板,30℃培养2天,挑取单菌落接种于装有50mlbmgy培养液(酵母提取物10g,胰蛋白胨20g,ynb13.4g,甘油10ml,1m磷酸钾(ph6.0)100ml,蒸馏水定容至1000ml)的三角瓶中,30℃,250rpm振荡培养至od600≈5.0。然后离心收集菌体,等量转移菌体沉淀至装有100mlbmmy培养液(酵母提取物10g,胰蛋白胨20g,ynb13.4g,甲醇10ml,1m磷酸钾(ph6.0)100ml,蒸馏水定容至1000ml)的三角瓶中28℃,250rpm振荡培养,每24小时添加1.5%的甲醇溶液进行诱导表达,诱导7天。发酵液5000rpm,4℃离心10min取得上清测定酶活;采用《sb/t10317-1999蛋白酶活力测定法》中的“福林法”测定sapp1的蛋白酶活。然后使用镍柱亲和层析纯化目的蛋白,其中镍柱亲和层析上样量为50ml,所用洗脱液为0.15m咪唑、0.5mnacl、0.02m磷酸缓冲液(ph7.4)。以转化空载体pich的gs115菌株作为实验对照。
通过前述方法,测得本发明所得的发酵液蛋白酶活为176.9u/ml,比酶活为1478.3u/mg。通过生工生物工程(上海)股份有限公司的bca蛋白浓度试剂盒测得纯化后的蛋白酶的产量为312.5mg/1000ml发酵液。
zuzanavinterová等在酿酒酵母里表达近平滑假丝酵母sapp1的产量为3mg/l。由此可见,本发明不仅成功异源表达纯化得到sapp1,且产量实现了明显提高。
以相似的重组表达方法对近平滑假丝酵母基因组内注释同为酸性蛋白酶的13个基因(对应的embl数据库的编号为cce40037,cce41985,cce45192,cce45175,cce45177,cce41276,cce40204,cce40220,cce39854,cce40477,cce40528,cce45016和cce45014))进行异源表达,通过福林酚法和水解圈法均无法检测到活性蛋白或蛋白活性非常低。
实施例3重组蛋白的sds-page检测
利用sds-page凝胶电泳来确认重组蛋白酶的表达情况、纯度和分子质量的大小。采用的浓缩胶浓度为12%以及分离胶浓度为5%,上样量为20μl,以标准分子量的标准蛋白作为marker。sds-page凝胶电泳的操作过程参见《蛋白质电泳实验技术》。对于发酵液样品的制备,诱导表达产生的重组蛋白酶的量较高,可以直接将发酵液稀释1倍后与上样缓冲液混匀,沸水煮沸10min后,12000rpm离心1min,上样后进行电泳。
粗酶液(指未经过镍柱亲和层析的发酵液)与纯化后酶液的sds-page电泳图如图2所示,m表示标准分子量蛋白,泳道1是经过7天发酵诱导后得到的粗酶液,泳道2是经过亲和层析纯化的样品,泳道3是经过7天诱导的转化空载体的毕赤酵母上清液。由图中可以看出,转化空载体的毕赤酵母并无蛋白表达,而未经纯化的阳性诱导(即泳道1)的上清液中有一深一浅两个蛋白条带,其中深色条带大小接近47kda,与预期结果相符;由泳道2可看出,经过纯化处理后只剩下单一的预期蛋白条带,表明已经成功获得电泳纯级别的酸性蛋白酶sapp1。
实施例4水解实验
配制蛋白水解平板(脱脂奶粉10g,琼脂粉20g,蒸馏水定容至1000ml),在平板上打孔,然后分别在孔内加入100μl的100u/ml的木瓜蛋白酶(北京奥博星生物技术有限公司,500000u/g)溶液,100u/ml的饲用酸性蛋白酶(宁夏夏盛实业集团有限公司,60000u/g)溶液及稀释至100u/ml实施例2得到的发酵液。在室温放置8小时后观察实验结果。以灭活的稀释至100u/ml的实施例2得到发酵液作为实验对照。
结果如图3所示,灭活的发酵液无水解圈,而在100u/ml的酶浓度下,本发明的发酵液的水解圈直径明显大于商业木瓜蛋白酶及饲用酸性蛋白酶,且水解圈更透明,表明该发酵液对脱脂奶粉蛋白的水解效果优于商业木瓜蛋白酶及饲用酸性蛋白酶。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
序列表
<110>华南理工大学
<120>近平滑假丝酵母酸性蛋白酶sapp1及表达、纯化方法
<160>8
<170>siposequencelisting1.0
<210>1
<211>376
<212>prt
<213>近平滑假丝酵母gim2.146(candidaparapsilosisgim2.146)
<400>1
ileprogluglualaalalysargaspaspasnproglyphevalala
151015
leuasppheaspvalleuarglysproleuasnleuthrglualaleu
202530
leuargglulysargaspserileserleuserleuileasnglugly
354045
prosertyralaserlysvalservalglyserasnlysglnglngln
505560
thrvalileileaspthrglyserseraspphetrpvalvalaspser
65707580
asnalaglncysglylysglyvalaspcyslysserserglythrphe
859095
thrprosersersersersertyrlysasnleuglyalaalaphethr
100105110
ileargtyrglyaspglyserthrserglnglythrtrpglylysasp
115120125
thrvalthrileasnglyvalserilethrglyglnglnilealaasp
130135140
valthrglnthrservalaspglnglyileleuglyileglytyrthr
145150155160
serasnglualavaltyraspthrserglyargglnthrthrproasn
165170175
tyraspasnvalprovalthrleulyslysglnglylysileargthr
180185190
asnalatyrserleutyrleuasnserproseralagluthrglythr
195200205
ileilepheglyglyvalaspasnalalystyrserglylysleuval
210215220
alagluglnvalthrserserglnalaleuthrileserleualaser
225230235240
valasnleulysglyserserpheserpheglyaspglyalaleuleu
245250255
aspserglythrthrleuthrtyrpheproseraspphealaalagln
260265270
leualaasplysalaglyalaargleuvalglnvalalaargaspgln
275280285
tyrleutyrpheileaspcysasnthraspthrserglythrthrval
290295300
pheasnpheglyasnglyalalysilethrvalproasnthrglutyr
305310315320
valtyrglnasnglyaspglythrcysleutrpglyileglnproser
325330335
aspaspthrileleuglyaspasnpheleuarghisalatyrleuleu
340345350
tyrasnleuaspalaasnthrileserilealaglnvallystyrthr
355360365
thraspserserileseralaval
370375
<210>2
<211>1128
<212>dna
<213>近平滑假丝酵母gim2.146(candidaparapsilosisgim2.146)
<400>2
attccagaggaggctgctaaaagagacgacaatcctgggtttgttgccttggactttgat60
gtgcttaggaaaccattgaacttgaccgaggcgcttctccgtgaaaagagagactccatt120
tcgttgtcgttgatcaatgaaggtccatcatatgcatctaaagtttcagtcggttcaaac180
aaacagcagcaaaccgtcattattgatactggttcgagtgacttttgggtagtggattca240
aatgcccaatgtggaaaaggtgttgattgcaagagctcagggacctttaccccatcatcg300
tcttcttcatacaagaatttaggagctgctttcactattcgatatggggatggatctaca360
tcgcaaggtacatggggtaaagataccgtcacaatcaatggagtctcaatcactggacaa420
caaattgcagatgtcactcaaacatcagttgatcaaggtatattgggtattggctacacc480
agcaatgaagcagtttacgacacgagtggtcgtcaaaccactccaaactacgacaacgtt540
cccgtgactttgaaaaagcaaggaaagatcagaaccaatgcttactcattatatttgaac600
tctccttcagctgagactggtacaatcatctttggtggtgttgataatgccaagtactct660
ggcaagttggttgccgagcaagttacctcttcacaagcgttgactatttcgcttgcttcc720
gtcaatttgaaaggttcatcattttcatttggagatggtgctttgttggactctggaacg780
acactcacttacttcccaagtgactttgccgcacaacttgctgataaagctggtgctcgt840
cttgttcaagtggctagagaccaatacttgtactttattgattgtaacaccgatacatct900
ggcacaacggtgtttaactttggaaatggggcaaagattaccgttccaaacacagagtac960
gtttatcaaaatggtgatggtacttgtctttggggtatccaaccatcagacgatactatt1020
ttaggtgacaactttttgagacatgcttacctcctttacaacttggatgcaaacacaatc1080
tcaatcgctcaagtcaagtacaccacagactcgagcatttctgccgtt1128
<210>3
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>3
gtcaggatccattccagaggaggctgctaa30
<210>4
<211>28
<212>dna
<213>人工序列(artificialsequence)
<400>4
atgcctgcagaacggcagaaatgctcga28
<210>5
<211>888
<212>dna
<213>人工序列(artificialsequence)
<400>5
agatctaacatccaaagacgaaaggttgaatgaaacctttttgccatccgacatccacag60
gtccattctcacacataagtgccaaacgcaacaggaggggatacactagcagcagaccgt120
tgcaaacgcaggacctccactcctcttctcctcaacacccactttaggctactaacacca180
tgactttattagcctgtctatcctggcccccctggcgaggttcatgtttgtttatttccg240
aatgcaacaagctccgcattacacccgaacatcactccagatgagggctttctgagtgtg300
gggtcaaatagtttcatgttccccaaatggcccaaaactgacagtttaaacgctgtcttg360
gaacctaatatgacaaaagcgtgatctcatccaagatgaactaagtttggttcgttgaaa420
tgctaacggccagttggtcaaaaagaaacttccaaaagtcgccataccgtttgtcttgtt480
tggtattgattgacgaatgctcaaaaataatctcattaatgcttagcgcagtctctctat540
cgcttctgaaccccggtgcacctgtgccgaaacgcaaatggggaaacacccgctttttgg600
atgattatgcattgtctccacattgtatgcttccaagattctggtgggaatactgctgat660
agcctaacgttcatgatcaaaatttcatgatcaaaatttaactgttctaacccctacttg720
acagcaatatataaacagaaggaagctgccctgtcttaaacctttttttttatcatcatt780
attagcttactttcataattgcgactggttccaattgacaagcttttgattttaacgact840
tttaacgacaacttgagaagatcaaaaaacaactaattattcgaaacg888
<210>6
<211>266
<212>dna
<213>人工序列(artificialsequence)
<400>6
atgagatttccatctattttcactgctgtattgtttgcagcatcttctgctttggctgct60
ccagttaacactactactgaagatgaaactgctcaaattccagctgaagctgttattggt120
tactctgatttggaaggtgattttgatgttgctgttttgccattttctaactctactaac180
aacggtttgttgtttatcaatactactattgcttcaattgctgctaaggaagaaggtgtt240
tctttggaaaagagagaagctgaggc266
<210>7
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>7
aacgttgttgccattgcggcaggcatcgtggtgtcacg38
<210>8
<211>71
<212>dna
<213>人工序列(artificialsequence)
<400>8
tacttgaagtcggacagtgagtgtagtcttgagaaattctgaagccgtatttttattatc60
agtgagtcagt71
- 还没有人留言评论。精彩留言会获得点赞!