一株生产凝胶态黄原胶的基因工程菌株及其构建方法和应用与流程
本发明属于工程菌的构建和应用技术领域,尤其涉及一株生产凝胶态黄原胶的基因工程菌及其构建方法和应用。
背景技术:
黄原胶是由野油菜黄单胞菌(xanthomonascampestris)产生的一种胞外阴离子多糖,是国际上集增稠、悬浮、乳化、稳定于一体,性能最优越的生物胶,已广泛应用于食品、农业、石油、印染及日化等行业。我国是黄原胶的重要生产基地,黄原胶产量约占全球总产量的67%。近几年全球对黄原胶的需求逐年上涨,我国黄原胶产量也一直呈现增长趋势。随着黄原胶应用领域的不断扩展以及下游产业对产品质量要求的提升,开发具有稳定优良特性的黄原胶产品是工业发展的一大趋势。
黄原胶具有类似纤维素的骨架结构,且在第二个葡萄糖上含有α-d-man-(2→1)-β-d-glca-(4→1)-β-d-man的侧链结构。根据不同的黄原胶生产菌株及发酵条件,接近90%的内侧甘露糖发生乙酰化,而30~50%的外侧甘露糖基团发生丙酮酸化。侧链上的乙酰基团可以稳定分子内部的螺旋构象,而丙酮酸基团可以促进黄原胶分子间的交联,因此,丙酮酸含量越高,黄原胶溶液的粘度越大。根据黄原胶的单体类型,丙酮酸含量在黄原胶分子中所占的最大比例为8.69%,而自然存在的黄原胶中丙酮酸含量一般低于5.0%。
黄原胶分子中丙酮酸和乙酰基含量的差异可显著影响黄原胶的流变学特性。在传统发酵过程中,由于发酵条件、出发菌株、发酵批次的不同,黄原胶分子中丙酮酸和乙酰基含量存在差异,进而影响了黄原胶的流变学特性。这一现状对黄原胶产品的质量分析、分选造成了困扰,并增加了黄原胶的生产成本及不稳定性。
技术实现要素:
有鉴于此,本发明的目的在于提供一株生产凝胶态黄原胶的基因工程菌及其构建方法和应用;所述基因工程菌产生的黄原胶具有饱和的丙酮酸含量,低浓度下即可形成弱凝胶,产品质量及流变学特性稳定。
为了实现上述发明目的,本发明提供了以下技术方案:
一株生产凝胶态黄原胶的基因工程菌,所述基因工程菌以xanthomonascampestrisnk-01为出发菌株,在所述出发菌株中敲除乙酰基转移酶ⅰ基因gumf和乙酰基转移酶ⅱ基因gumg,并且过表达酮基转移酶基因guml;
所述xanthomonascampestrisnk-01的菌种保藏编号为cgmccno.15155。
优选的,所述过表达酮基转移酶基因guml是通过向所述出发菌株中转入包含酮基转移酶基因guml的多拷贝表达质粒pbbrl实现。
本发明提供了所述的基因工程菌的制备方法,包括以下步骤:
1)敲除所述出发菌株中的gumf基因和gumg基因,获得gumf基因和gumg基因缺失的菌株xc(△fg);
2)在步骤1)中所述的gumf基因和gumg基因缺失的菌株xc(△fg)中过表达酮基转移酶基因guml获得基因工程菌。
优选的,步骤1)中敲除所述出发菌株中的gumf基因和gumg基因包括以下步骤:
1.1)以所述出发菌株的基因组为模板,进行pcr扩增获得基因gumf的上游同源臂片段和基因gumg的下游同源臂片段;所述基因gumf的上游同源臂片段的核苷酸序列如seqidno:1所示;所述基因gumg的下游同源臂片段的核苷酸序列如seqidno:2所示;
1.2)将步骤1)获得的所述基因gumf的上游同源臂片段和基因gumg的下游同源臂片段通过overlappcr重组后,连接至自杀质粒plo3中,获得无痕敲除质粒plo3-fg;
1.3)将步骤2)中所述无痕敲除质粒plo3-fg转入出发菌株中进行gumf和gumg基因的敲除获得gumf和gumg基因缺失的菌株xc(△fg)。
优选的,步骤2)中所述过表达酮基转移酶基因guml包括以下步骤:
2.1)将酮基转移酶基因guml与载体pbbr1mcs-2连接获得多拷贝表达质粒pbbrl;
2.2)将步骤2.1)中所述多拷贝表达质粒pbbrl转入到所述gumf和gumg基因缺失的菌株xc(△fg)中获得基因工程菌xc(△fg::pbbrl)。
优选的,步骤1.3)中所述的转入为接合转移,所述出发菌株和无痕敲除质粒plo3-fg的数量比为1:(1.5~2.5);所述接合转移的温度为28~32℃,所述接合转移的时间为8~12h。
本发明提供了一种利用所述基因工程菌生产黄原胶的方法,包括以下步骤:
s1)将所述基因工程菌接种到种子培养基中进行活化获得活化菌液;
s2)将所述活化菌液接种到发酵培养基中进行发酵培养获得发酵液;所述发酵培养的温度为28~32℃,所述发酵培养的时间为60~84h;
s3)醇沉所述发酵液,收集固相组分为黄原胶。
优选的,步骤s1)中所述活化菌液的菌浓度od600为0.4~0.6。
优选的,步骤s2)中所述活化菌液的接种量为8~12%(v/v)。
优选的,所述发酵培养的培养基以1l计,包括以下组分:玉米淀粉,40~60g;鱼蛋白胨,3.5~4.5g;碳酸钙,3.5~4.5g和余量的水。
本发明的有益效果:本发明所述生产凝胶态黄原胶的基因工程菌,以xanthomonascampestrisnk-01为出发菌株,敲除了所述出发菌株中的乙酰基转移酶ⅰ基因gumf和乙酰基转移酶ⅱ基因gumg,并过表达了酮基转移酶基因guml;所述基因工程菌生产的黄原胶具有更高的丙酮酸含量,接近于饱和;在低浓度条件下即为弱凝胶,随着浓度的增加,零剪切粘度和储能模量逐渐增加,当达到2%浓度后,黄原胶呈现较强的凝胶态,具有一定的自持性。所述基因工程菌生产的黄原胶在水溶液或者盐溶液中均呈现凝胶态,凝胶强度随着黄原胶浓度和离子浓度的增加而增加;比自然产生的黄原胶具有更加优越的增稠和凝胶特性,且不受发酵条件的限制,可稳定高效生产。因此,本发明所述基因工程菌株具有广泛的工业应用前景。
附图说明
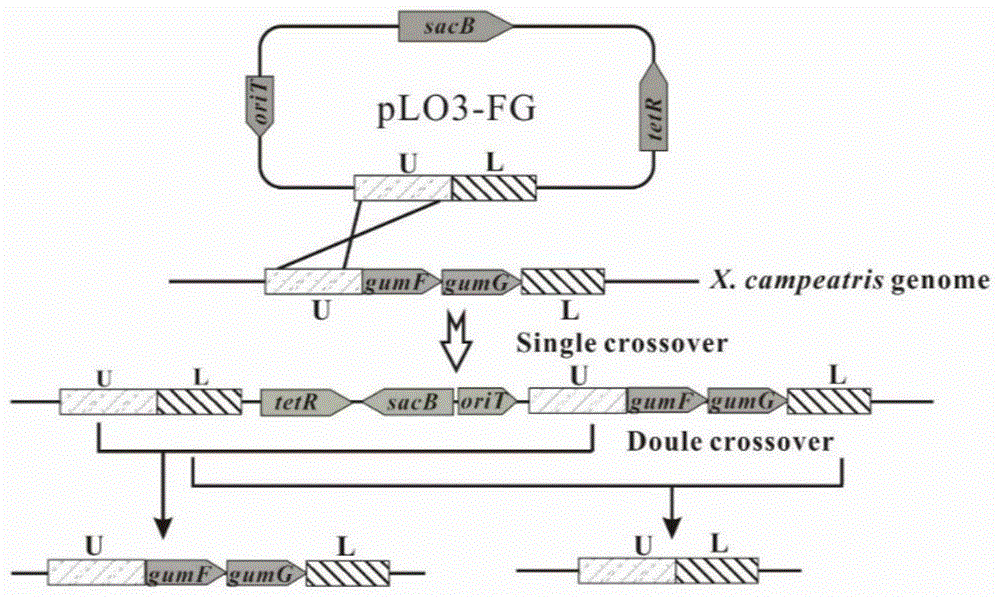
图1为本发明实施例1中gumf和gumg基因的敲除流程图;
图2为缺失菌株的pcr验证图,其中m为dnamarkerdl2000,泳道w为出发菌株的pcr结果,泳道δ为gumf和gumg基因缺失的菌株xc(△fg)的pcr结果;
图3为实施例3不同黄原胶产品的粘度随剪切力变化曲线图;
图4为实施例3中不同浓度的黄原胶xc-l的粘度(a)和模量(b)变化曲线;
图5为黄原胶xc-l在不同nacl浓度下的粘度(a)和模量(b)变化曲线;
图6为黄原胶xc-l的凝胶态观察,a指不同浓度的xc-l的凝胶态,b指1.0%的xc-l在不同浓度na+中的凝胶态;照片于倒置12小时后拍摄;
图7为黄原胶xc-l(a)和xc-5(b)在nacl溶液中的显微结构及高度线图,a图中1和2代表多螺旋链,b图中1-3代表双螺旋链;
图8为质粒plo3的质粒图谱。
生物保藏说明
野油菜黄单胞菌xanthomonascampestrisnk-01保藏于中国普通微生物菌种保藏中心,保藏地址为北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所;保藏日期为2018年01月03日,保藏编号为cgmccno.15155。
具体实施方式
本发明提供了一株生产凝胶态黄原胶的基因工程菌,所述基因工程菌以xanthomonascampestrisnk-01为出发菌株,在所述出发菌株中敲除乙酰基转移酶ⅰ基因gumf和乙酰基转移酶ⅱ基因gumg,并且过表达酮基转移酶基因guml;所述xanthomonascampestrisnk-01的菌种保藏编号为cgmccno.15155。
本发明中,优选的通过将连接有基因gumf的上游同源臂序列和基因gumg的下游同源臂序列的无痕敲除质粒转入出发菌株xanthomonascampestrisnk-01中进行基因gumf和基因gumg的敲除。本发明中,所述过表达酮基转移酶基因guml优选的是通过向所述出发菌株中转入包含酮基转移酶基因guml的多拷贝表达质粒pbbrl实现。
本发明中所述基因工程菌敲出了乙酰基转移酶ⅰ基因gumf和乙酰基转移酶ⅱ基因gumg;并且过表达了酮基转移酶基因guml;使得所述基因工程菌在合成黄原胶的过程中,降低黄原胶中乙酰基的含量,提高黄原胶中丙酮酸的含量;从而使得所述基因工程菌能够生产凝胶态黄原胶。本发明所述基因工程菌合成的黄原胶具有饱和的丙酮酸含量,低浓度下即可形成弱凝胶,产品质量及流变学特性稳定,有利于黄原胶产品的定向稳定生产。
本发明还提供了基因工程菌的制备方法,包括以下步骤:1)敲除所述出发菌株中的gumf基因和gumg基因,获得gumf基因和gumg基因缺失的菌株xc(△fg);2)在步骤1)中所述的gumf基因和gumg基因缺失的菌株xc(△fg)中过表达酮基转移酶基因guml获得基因工程菌。
在本发明中,所述出发菌株中的gumf基因和gumg基因优选的包括以下步骤:1.1)以所述出发菌株的基因组为模板,进行pcr扩增获得基因gumf的上游同源臂片段和基因gumg的下游同源臂片段;所述基因gumf的上游同源臂片段的核苷酸序列如seqidno:1所示;所述基因gumg的下游同源臂片段的核苷酸序列如seqidno:2所示;1.2)将步骤1)获得的所述基因gumf的上游同源臂片段和基因gumg的下游同源臂片段通过overlappcr重组后,连接至自杀质粒plo3中,获得无痕敲除质粒plo3-fg;1.3)将步骤2)中所述无痕敲除质粒plo3-fg转入出发菌株中进行gumf和gumg基因的敲除获得gumf和gumg基因缺失的菌株xc(△fg)。
本发明以所述出发菌株的基因组为模板,进行pcr扩增获得基因gumf的上游同源臂片段和基因gumg的下游同源臂片段。本发明对所述出发菌株的基因组的提取方法没有特殊限定,采用本领域常规的基因组提取方法即可,在本发明具体实施过程中,采用基因组试剂盒提取。本发明中,扩增所述基因gumf的上游同源臂片段的引物优选为fgsu和fgsl;所述fgsu的核苷酸序列如seqidno:3所示;所述fgsl的核苷酸序列如seqidno:4所示;扩增所述基因gumg的上游同源臂片段的引物优选为fgxu和fgxl;所述fgxu的核苷酸序列如seqidno:5所示;所述fgxl的核苷酸序列如seqidno:6所示。本发明中所述pcr扩增所用的酶优选为primestardna聚合酶;本发明中,所述pcr扩增的体系以25μl计,优选的包括以下组分:
出发菌株的基因组dna,1μl;
引物fgsu和fgsl(或者引物fgxu和fgxl),各0.4μl;
primestardna聚合酶,12.5μl;
余量的水。
所述pcr扩增的程序优选的包括:98℃,15s;55℃,15s;72℃,40s;30循环;16℃保持。
本发明在获得所述基因gumf的上游同源臂片段和基因gumg的下游同源臂片段后,通过overlappcr重组上述片段,然后将重组的片段连接至自杀质粒plo3中,获得无痕敲除质粒plo3-fg。
在本发明中,所述overlappcr扩增体系以25μl计,包括以下组分:
上下游扩增片段,各0.5μl;
引物fgsu,fgxl,各0.3μl;
primestardna聚合酶,12.5μl;
余量的水。
所述pcr扩增的程序优选的包括:98℃,15s;55℃,15s;72℃,40s;30循环;16℃保持。
本发明获得重组的片段后,优选的将所述重组的片段和自杀质粒用相同的酶进行酶切,然后连接获得无痕敲除质粒plo3-fg。本发明中,所述酶切优选为双酶切;所述双酶切用酶优选为限制性内切酶pacⅰ和xbaⅰ;所述双酶切的温度优选为35~39℃,更优选为37℃,所述双酶切的时间优选为1.5~2.5h,更优选为2h。
所述酶切体系以10μl计,包括以下组分:
重组片段或质粒,7μl;
限制性内切酶pacⅰ和xbaⅰ,各1μl;
酶切buffer,1μl。
本发明中,所述酶切结束后,优选的收集酶切后的重组片段和酶切后的质粒进行连接;所述连接的温度优选为16℃,所述连接的时间优选为10~14h。本发明在所述连接后,优选的将所述连接产物转入大肠杆菌感受态中获得带有无痕敲除质粒plo3-fg的重组菌;本发明优选的采用菌落pcr验证,筛选正确的转化子。本发明对所述菌落pcr验证的具体操作没有特殊限定采用本领域常规的菌落pcr验证方法即可;在本发明具体实施过程中,所述菌落pcr验证的引物为fg1和fg2;所述fg1的核苷酸序列如seqidno:7所示;所述fg2的核苷酸序列如seqidno:8所示。本发明中,所述自杀质粒plo3的质粒图谱如图8所示。
本发明在获得所述无痕敲除质粒plo3-fg后,将所述无痕敲除质粒plo3-fg转入出发菌株中进行gumf和gumg基因的敲除获得gumf和gumg基因缺失的菌株xc(△fg)。在本发明中,所述的转入优选为接合转移,所述出发菌株和无痕敲除质粒plo3-fg的数量比优选为1:(1.5~2.5),更优选为1:2;所述接合转移的温度优选为28~32℃,更优选为30℃;所述接合转移的时间优选为8~12h,更优选为9~11h。本发明中,优选的将带有无痕敲除质粒plo3-fg的重组菌与出发菌株进行接合转移。
在本发明具体实施过程中,优选的将所述出发菌株与带有无痕敲除质粒plo3-fg的重组菌分别置于含cm抗性和含tet抗性的液体培养基中进行培养后,收集菌体;将所述收集到的菌体洗涤重悬后,混合获得混合菌,将所述混合菌置于无抗性固体培养基中进行接合转移。本发明中,所述洗涤重悬用的溶液优选为mgso4溶液,所述mgso4溶液的浓度优选为8~12mmol/l。本发明中,所述无抗性固体培养基中优选的含有滤膜,所述滤膜有利于菌体的分离。本发明在所述接合转移后,优选的从所述滤膜上洗涤菌体,然后将从滤膜上洗涤下来的菌体涂布含有cm和tet的双抗的培养基上进行筛选和验证。本发明优选的采用菌落pcr验证,筛选正确的转化子。本发明对所述菌落pcr验证的具体操作没有特殊限定采用本领域常规的菌落pcr验证方法即可;在本发明具体实施过程中,所述菌落pcr验证的引物为fg1和fg2。
本发明中,在所述的gumf基因和gumg基因缺失的菌株xc(△fg)中过表达酮基转移酶基因guml获得基因工程菌。所述过表达酮基转移酶基因guml优选的包括以下步骤:2.1)将酮基转移酶基因guml与载体pbbr1mcs-2连接获得多拷贝表达质粒pbbrl;2.2)将步骤2.1)中所述多拷贝表达质粒pbbrl转入到所述gumf和gumg基因缺失的菌株xc(△fg)中获得基因工程菌xc(△fg::pbbrl)。
本发明中,优选的以出发菌株的基因组为模板进行pcr扩增获得所述酮基转移酶基因guml。本发明对所述出发菌株的基因组的提取方法没有特殊限定,采用本领域常规的基因组提取方法即可,在本发明具体实施过程中,采用基因组试剂盒提取。本发明中,扩增所述基因guml的引物优选为l-br1和l-br2;所述l-br1的核苷酸序列如seqidno:9所示;所述l-br2的核苷酸序列如seqidno:10所示。本发明中,所述pcr扩增的体系以25μl计,优选的包括以下组分:
出发菌株的基因组dna,1μl;
引物l-br1和l-br2,各0.4μl;
primestardna聚合酶,12.5μl;
余量的水。
所述pcr扩增的程序优选的包括:98℃,15s;55℃,15s;72℃,40s;30循环;16℃保持。
本发明在获得所述酮基转移酶基因guml后,将酮基转移酶基因guml与载体pbbr1mcs-2连接获得多拷贝表达质粒pbbrl。本发明中,所述酶切优选为双酶切;所述双酶切用酶优选为限制性内切酶kpnⅰ和hindⅲ;所述双酶切的温度优选为35~39℃,更优选为37℃,所述双酶切的时间优选为1.5~2.5h,更优选为2h。所述酶切体系以10μl计,包括以下组分:
重组片段或质粒,7μl;
限制性内切酶kpnⅰ和hindⅲ,各1μl;
酶切buffer,1μl。
本发明中,所述酶切结束后,优选的收集酶切后的基因guml和酶切后的质粒进行连接;所述连接的温度优选为16℃,所述连接的时间优选为10~14h。本发明在所述连接后,优选的将所述连接产物转入大肠杆菌感受态中获得带有多拷贝表达质粒pbbrl的重组菌;本发明优选的采用菌落pcr验证,筛选正确的转化子。
本发明在获得所述多拷贝表达质粒pbbrl后,将所述多拷贝表达质粒pbbrl转入到所述gumf和gumg基因缺失的菌株xc(△fg)中获得基因工程菌xc(△fg::pbbrl)。本发明中所述转入与上述将无痕敲除质粒plo3-fg转入出发菌株中的操作一致,在此不再赘述。
本发明提供了一种利用所述基因工程菌生产黄原胶的方法,包括以下步骤:s1)将所述基因工程菌接种到种子培养基中进行活化获得活化菌液;s2)将所述活化菌液接种到发酵培养基中进行发酵培养获得发酵液;所述发酵培养的温度为28~32℃,所述发酵培养的时间为60~84h;s3)醇沉所述发酵液,收集固相组分为黄原胶。
在本发明中,将所述基因工程菌接种到种子培养基中进行活化获得活化菌液。在本发明中,所述活化优选的包括1~3代活化,更优选的包括2代活化。本发明中所述活化菌液的菌浓度od600优选为0.4~0.6,更优选为0.5。本发明中,所述种子培养基以1l计优选的包括以下组分:蔗糖,15~25g;蛋白胨,2.5~3.5g;酵母粉,0.8~1.2g;牛肉膏,4~6g;更优选为包括以下组分:蔗糖,20g;蛋白胨,3g;酵母粉,1g;牛肉膏,5g;所述种子培养基的ph值优选为6.98~7.02。
本发明在获得活化菌液后,将所述活化菌液接种到发酵培养基中进行发酵培养获得发酵液。所述活化菌液的接种量优选为8~12%(v/v),更优选为10%(v/v)。本发明中,所述发酵培养的培养基以1l计,优选的包括以下组分:玉米淀粉,40~60g;鱼蛋白胨,3.5~4.5g;碳酸钙,3.5~4.5g和余量的水;更优选为包括以下组分:玉米淀粉,50g;鱼蛋白胨,4g;碳酸钙,4g和余量的水。所述发酵培养的温度为28~32℃,优选为30℃,所述发酵培养的时间优选为60~84h,更优选为65~80h,最优选为72h。本发明中,所述活化和培养的过程中,优选的伴随搅拌,所述搅拌的转速优选为150~250rpm,更优选为200rpm。
本发明在所述发酵结束后,醇沉所述发酵液,收集固相组分为黄原胶。本发明中,所述醇沉所用的醇优选为乙醇,所述乙醇与发酵液的体积比优选为(2~3):1;所述醇沉过程中优选的伴随搅拌,本发明对所述搅拌的转速没有特殊限定。本发明在所述醇沉结束后,优选的采用过滤的方法收集固相组分获得黄原胶。本发明在获得所述黄原胶后优选的还包括干燥和粉碎的步骤,所述干燥的温度优选为85~95℃,更优选为90℃;所述干燥的时间优选为4~8h,更优选为6h;所述粉碎的粒度优选为60~100目,更优选为80目。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
本发明实施例中使用的引物序列见表1。
表1、引物序列信息表
实施例1
无痕敲除质粒plo3-fg的构建
利用基因组试剂盒提取xc的基因组,以其为模板,以fgsu/fgsl和fgxu/fgxl引物,通过primestardna聚合酶(takarabio,tokyo,japan)分别扩增gumf基因上游同源臂片段和gumg基因的下游同源臂片段;overlappcr重组上下游片段,琼脂糖凝胶电泳分离、胶回收试剂盒回收目的片段。用限制性内切酶pacⅰ和xbaⅰ,37℃,2h分别酶切重组目的片段及质粒plo3(来源于oliverlenz博士,质粒图谱如图8所示),16℃连接过夜,转化至e.colis17感受态细胞中;利用引物fg1/fg2进行菌落pcr验证以筛选正确的转化子,并保存甘油管备用。
基因gumf和基因gumg的pcr及扩增体系如下:
pcr体系25μl:模板1μl,
引物各0.4μl,
酶12.5μl,
水补齐。
所述overlappcr扩增体系如下:
上下游扩增片段,各0.5μl;
引物fgsu,fgxl,各0.3μl;
primestardna聚合酶,12.5μl;
余量的水。
基因gumf和基因gumg的pcr及两个基因overlappcr扩增程序如下:
98℃15s
55℃15s
72℃40s30循环
16℃。
基因gumf和gumg的敲除流程如图1所示。
广宿主范围表达载体pbbrl的构建
利用基因组试剂盒提取xc的基因组,以其为模板,以l-br1/l-br2为引物,通过primestardna聚合酶(takarabio,tokyo,japan)扩增guml基因;用限制性内切酶kpnⅰ和hindⅲ,37℃,2h分别酶切guml及质粒pbbr1mcs-2,16℃连接过夜,转化至e.colis17感受态细胞中;利用引物l-br1/l-br2进行菌落pcr验证以筛选正确的转化子,并保存甘油管备用。
基因guml的pcr及扩增体系如下:
pcr体系25μl:模板1μl,
引物各0.4μl,
酶12.5μl,
水补齐。
扩增程序:
98℃15s
55℃15s
72℃40s30循环
16℃
菌落pcr程序:
95℃45s
55℃45s
72℃2min30循环
16℃。
凝胶态黄原胶的基因工程菌株xc(△fg::pbbrl)的构建
挑xc单菌落接种至含cm的5ml种子培养基,30℃恒温摇床中200rpm培养15h,挑取e.colis17/plo3-fg的单菌落至含有tet的lb液体培养基中,于37℃恒温摇床中200rpm培养8h,6000rpm离心5min收集两株菌的菌体,10mmol/l的mgso4溶液洗涤菌体两次,200μl的mgso4溶液重悬菌体,将xc和e.colis17(plo3-fg)按照1:2的比例混匀后放置在含0.22μm滤膜的无抗性固体培养基上,30℃条件下培养8~12h。用200μlmgso4溶液洗涤滤膜,梯度稀释后涂布含有cmr和tetr的双抗平板上,30℃、48~72h培养。plo3在xc中无法复制,当plo3-fg接合转移至xc后,其会通过上游同源臂或下游同源臂重组整合入基因组。在双抗平板上挑取单菌落至含双抗的种子培养基中,30℃、200rpm培养36h获得单交换重组子。随后,将单交换重组子接种至无抗性的种子培养基中,30℃、200rpm培养24h,传代两次,然后将其涂布至含10%蔗糖的平板上,30℃条件下培养72h。挑取单菌落至5ml试管中,30℃,200rpm培养24h,使用引物fg1/fg2进行菌落pcr验证,确定正确的缺失菌株,将其命名为xc(△fg)。利用相同的方法,以xc(△fg)为受体菌株,以e.colis17(pbbrl)为供体菌株进行接合转移,构建可产生凝胶态黄原胶的基因工程菌株,命名为xc(△fg::pbbrl),其发酵产生的黄原胶命名为xc-l。
其中无抗性固体培养基组分为(g/l):
葡萄糖:15g,牛肉膏:3g,蛋白胨:5g,酵母粉:3g,琼脂:15g,ph=7。
上述步骤中涉及到的菌落pcr的程序如下:
95℃45s
55℃45s
72℃2min30循环
16℃。
缺失菌株的pcr验证结果入图2所示,其中m为dnamarkerdl2000,泳道w为出发菌株的pcr结果,泳道δ为gumf和gumg基因缺失的菌株xc(△fg)。
实施例2
实施例1中构建的基因工程菌发酵生产凝胶态黄原胶
挑基因工程菌单菌落接种到5ml种子培养基,200rpm、30℃培养20h,然后以1%的接种量接种到100ml的种子培养基中使其菌浓od600达到0.5,在200rpm、30℃培养18h,然后以10%的接种量接种到含100ml培养基的500ml摇瓶中,30℃、200rpm条件下培养72h。
其中,种子培养基(g/l):蔗糖,20g;蛋白胨,3g;酵母粉,1g;牛肉膏,5g;ph7.0±0.02;发酵培养基(g/l):玉米淀粉,50g;鱼蛋白胨,4g;碳酸钙,4g。
向发酵液中加入2~3倍体积的乙醇(v/v),搅拌,获得黄原胶沉淀物,过滤,将沉淀放入烘箱90℃干燥6h,粉碎至粒径为80目,保存备用。
实施例3
基因工程菌生产的黄原胶的性能研究
黄原胶中乙酰基和丙酮酸含量的测定
乙酰基和丙酮酸的含量通过高压液相色谱仪(hplc,aglient1100,usa)测定。将黄原胶溶于水配置为5mg/ml的黄原胶水溶液,90℃加热搅拌1h,超声2min后溶液保存备用。丙酮酸含量测定方法如下:取1ml黄原胶溶液与1ml0.1mol/l的磷酸溶液混合,90℃下恒温90min,加水补足3ml,获得丙酮酸测定液;乙酰基含量测定方法如下:取1ml黄原胶溶液与1ml0.2mol/l的氢氧化钾溶液混合,充氮气,密封,45℃恒温6h后,用1ml0.1mol/l的磷酸溶液补足3ml,得到乙酰基测定液。在丙酮酸和乙酰基测定液中加入等体积的0.7g/l的丙酸为内标,过滤,取20μl进样进行hplc分析。丙酮酸含量标准曲线测定所用丙酮酸浓度为:15,30,45,75,100mg/ml;乙酰基含量标准曲线所用乙酸浓度为:15,30,45,75,150mg/ml。不同来源黄原胶的测定结果见表2。
表2不同来源黄原胶产品的乙酰基和丙酮酸含量
其中a表示xc菌株的不同发酵条件。xc-l、xc-2和xc-3的发酵条件为28℃,转速200rpm,发酵72h;xc-4发酵条件为33℃,转速100rpm,发酵72h;xc-4发酵条件为37℃,转速100rpm,发酵72h。
黄原胶溶液的流变学行为评估
黄原胶的流变学测定利用应力调节的流变仪(discoveryhr-2hybridrheometer,tainstruments),安装有直径40mm的铝板(gap1000μm)。粘度测定参数为:转速0.01-101/s,温度25℃;模量测定参数为:应变5%,温度25℃,频率0.1-100rad/s。储能模量一般代表样品的凝胶强度。黄原胶溶液配制浓度为0.1-2.0%,v/v;1.0%的黄原胶溶盐溶液中含有的氯化钠浓度为1-5000mm。
测定结果见表3和表4。
表3不同的黄原胶产品(0.3%)的流变学特性
表4基因工程菌株xc(△fg::pbbrl)产生黄原胶xc-l的流变学特性
其中a指1%的黄原胶溶解于含不同浓度的nacl溶液中。具体浓度为1、10、100、1000、3000、5000mm的nacl溶液。
不同黄原胶产品的粘度随剪切力变化曲线如图3所示。
不同浓度的黄原胶xc-l的粘度(a)和模量(b)变化曲线如图4所示;
黄原胶xc-l在不同nacl浓度下的粘度(a)和模量(b)变化曲线如图5所示。
黄原胶的三维结构观察
将黄原胶溶于纯水或者5000mm氯化钠溶液中,使其浓度为1.0mg/ml,过夜溶解。用相同的溶剂稀释至终浓度10μg/ml,取2μl黄原胶溶液置于新鲜的云母表层,室温干燥。用原子力显微镜(afm,burkericondimension)观察,nanoscopeanalysis1.8软件分析黄原胶分子的高度,其高度代表了黄原胶分子的交联程度,以此判断黄原胶分子的三维结构。高度越高,交联分子链越多;高度越低,交联分子链越少。结果如表5所示。
表5黄原胶产品xc-l与xc-5的分子链高度比较
黄原胶xc-l的凝胶态观察如图6所示,a指不同浓度的xc-l的凝胶态,b指1.0%的xc-l在不同浓度na+中的凝胶态;照片于倒置12h后拍摄。
黄原胶xc-l(a)和xc-5(b)在nacl溶液中的显微结构及高度线图如图7所示,其中a图中1和2代表多螺旋链,b图中1-3代表双螺旋链。
由表2可知,由基因工程菌株xc(δfg::pbbrl)产生的黄原胶xc-l具有更高的丙酮酸含量,接近于饱和;而野生型xc在不同发酵条件下产生的黄原胶产品的丙酮酸含量不同且较低。表3显示黄原胶xc-l具有更高的零剪切粘度和储能模量,且储能模量大于损耗模量,由此可知,xc-l在低浓度条件下即为弱凝胶。表4显示基因工程菌株xc(△fg::pbbrl)产生黄原胶xc-l溶于水后,随着黄原胶浓度的增加,零剪切粘度和储能模量逐渐增加,当达到2%浓度后,黄原胶呈现较强的凝胶态,具有一定的自持性。同样地,在nacl溶液中,随着na+离子浓度的增加,零剪切粘度和储能模量逐渐增加,当na+浓度达到5000mm时,零剪切粘度和储能模量急剧上升,说明xc-l在高盐溶液中具有很高的凝胶强度。为了解释xc-l与其它黄原胶的差异,以及xc-l的凝胶机制,对两种黄原胶产品进行了原子力显微镜观察,结果发现:在水溶液中,xc-l和xc-5均存在单链和双链螺旋,但xc-l的高度稍高于xc-5,说明xc-l可能具有较弱的分子内部相互作用力和较强的分子间相互作用力;在盐溶液中,xc-l表现为多股螺旋,而xc-5以双螺旋链形式存在,说明xc-l在盐溶液中的分子间作用力得到了进一步加强,进而呈现凝胶态。
综上所述,基因工程菌株xc(△fg::pbbrl)产生黄原胶xc-l在水溶液或者盐溶液中均呈现凝胶态,凝胶强度随着黄原胶浓度和离子浓度的增加而增加。比自然产生的黄原胶具有更加优越的增稠和凝胶特性,且不受发酵条件的限制,可稳定高效生产。因此,该基因工程菌株具有广泛的工业应用前景。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>南开大学
<120>一株生产凝胶态黄原胶的基因工程菌株及其构建方法和应用
<160>10
<170>siposequencelisting1.0
<210>1
<211>969
<212>dna
<213>人工序列(artificialsequence)
<400>1
atggcgatctggtcaacccgaagagcttcttcgtcaatgcgcgcggcatgagtgcagaag60
ggttctggaacgaggacagcaatctgttcgtcagtgccacacgacccggtgagcgcaact120
tcctcccaggctcgaacctgccacgcgcctcttcctggttcatcgagccggtgacgatgg180
gcaattacatctgcttcttcaccgcgatcgtattgacgttctggcgctggatgcggccgt240
cgatgctgattctgtctattggattgatcggcttcatgattgtggcatccgacggccgac300
tggctgccggcacctgtgtgctgatggtgctgctgtcgccgttattgaaacggatggatc360
agcggttggcgttcctgttgttcctgtttgtgatcgcctctgcctggctgctggtgtgga420
tgaccgggattacggcctaccaggacaccacgatggggcgcatcttcttcactgtgaatt480
cgatgaacaatctatcgttcgagtcgtggatgggcctggattttgcgcaggcctaccggt540
atttcgacagcggtatttcttactttattgcttcgcagtcgattgtcggcgtgctggcgt600
tcctgctgtcttattcgttcctgctgctgatgccgagcaaggaagggcagttgttcaaaa660
accaggcgatgtttgcctttgcactgagcctgttggtgtctaacggctatttctcgatca720
agacatcggcgctgtggtggtttgtctgcggctgcatgtggcacctgatgccagcagcgt780
cagccgtgccggtgcgcgacgaaagcaaggaagatccaacggacaacggcgtgcatgtgc840
cgttgcccgcaggagcgccgcggtgaatacggtgacaggggcatcggggacgtcggcgcc900
tgtgcaggctgccggcgcgcgtgccttcgcgagcggccgtagccgcgatccacgtatcga960
tgcgaccaa969
<210>2
<211>977
<212>dna
<213>人工序列(artificialsequence)
<400>2
ggtgatcgatcgcaaactcatcggtacacctgtctgggcgctggctctctgcgcctttgc60
catcgctgcctgcattcccatgcgtgccgtgctggtgcgccgcgccctggatgttgggat120
tgaaacgcaagtgagacattttcagaatcatcagtcgatgtggcgtgttcgtgtgagtca180
ccggcaaaggagatcggcgcaatgaaagtcgtgcatgtggtccgccagttccatccgtcg240
atcggggggatggaggaagtcgtgctgaacgtggcacgtcagcatcaggccaacagtgcc300
gacacggttgagatcgtgacgttggatcgtgtgttcaccgatccctctgcgcaactggcg360
cagcacgagctccatcaggggttgtcgatcactcgcatcggctatcgtggttcatcgcgg420
tacccgatcgcgccgtcggtgctgggggcgatccgttcggcggacgtggtgcatctgcat480
ggcattgattttttctacgactacctggcgttgaccaagccgctgcacggcaagccgatg540
gtggtctcgacgcatggcgggtttttccacactgcctatgcgtcgcgcatgaagcagatc600
tggttccagacgctgacgcgtacttctgcgctggcctatgcgcgtgtgatcgccactagc660
gagaatgacggcgatctgttcgccaaggtggtcgcgccgtcgcgcttgcgggtgatcgag720
aacggtgtcgacgtggagaagtatgcagggcagggcgctcgagcgccgggacggaccatg780
ctgtatttcgggcgttggtcggtcaacaagggcctgatcgaaacgcttgaattgctgcag840
gctgcgctcacgcgtgatccgcagtggcggttgatcatcgccgggcgcgagtacgatttg900
aatgaggcggatctgcgcaaggccatcgccgaacgcggtttgcaggacaaggtgcagctg960
agcatgtcgccatcgca977
<210>3
<211>34
<212>dna
<213>人工序列(artificialsequence)
<400>3
ggagttgttaattaaatggcgatctggtcaaccc34
<210>4
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>4
gagtttgcgatcgatcaccttggtcgcatcgatacgtg38
<210>5
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>5
cacgtatcgatgcgaccaaggtgatcgatcgcaaactc38
<210>6
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>6
gacatagtctagatgcgatggcgacatgctc31
<210>7
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>7
cggtatttcgacagcggtatt21
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>8
tctcctttgccggtgactca20
<210>9
<211>33
<212>dna
<213>人工序列(artificialsequence)
<400>9
gaaatgaggtacccgtttgaaggaggatccctg33
<210>10
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>10
gaacattaagcttgaaggcttcctgcgtggtc32
- 还没有人留言评论。精彩留言会获得点赞!