一种岷江百合LrWRKY2基因及应用的制作方法
本发明属于植物基因工程技术领域,特别的涉及一种岷江百合lrwrky2基因及应用。
背景技术:
株型是观赏植物的重要性状之一,选育理想株型是观赏植物育种的一个重要目标。匍匐生长的植物因其植株低矮、分枝角度小,贴近地面生长,因此更适合被用于地被景观的营造,在园林中有广阔的应用前景。研究匍匐型的转基因植物有助于扩大其应用价值,进一步推广其在园林绿化造景中的应用。
wrky是植物体内广泛存在的且保守的一类转录因子家族。最早被鉴定的植物wrky基因是甘薯(impoeabatatas)spf1(spf1:sweetpotatofactor1)基因,其基因产物特异地与甘薯sporamin基因和β-淀粉酶基因启动子区域的sp8序列识别,从而参与并调控植物糖信号途径的建立。目前wrky基因在多数植物中被相继发现,如拟南芥中已经发现74个wrky成员,在水稻中发现了109个wrky成员;wrky转录因子包括3个结构域,即dna结合域、核定位信号和寡聚化位点。wrky转录因子最主要的结构特点是各成员的dna结合域中都至少含有一个wrky结构域。wrky结构域是一段大约由60个氨基酸残基所组成的多肽序列,其中wrkygqk为所有成员中高度保守的7个氨基酸残基。除上述比较保守的区域外,wrky成员中其余氨基酸组成的同源性并不高。此外,wrky转录因子的dna结合域中一般还含有一个锌指结构。
wrky转录因子在转录水平上对靶基因进行调控,wrky蛋白大部分定位与细胞核,对一些wrky蛋白氨基酸序列分析发现,在wrky结构域以外存在细胞核定位信号区(nls),这意味着wrky蛋白在胞质中合成后,通过跨膜运输进入细胞核发挥作用。体外瞬间实验也证明了一些存在nls的wrky蛋白定位于细胞核。wrky转录因子通过结合靶基因启动子区域w盒(c/t)tgac(t/c)核苷酸序列而调控相应基因表达。研究表明,wrky转录因子是植物对病原体防卫反应的关键组成部分,在植物防卫反应中起重要作用。已证实wrky转录因子参与植物的损伤与衰老反应、参与植物生长发育代谢过程以及响应植物的生物胁迫和各种非生物胁迫,如高盐、高温、干旱和寒冷等。如在hsp101启动子的驱动下过表达oswrky11,通过使植物减慢萎蔫速率和增加逆境存活率从而表现出对干旱的耐受性(wux.,etal.,plantcellrep.2009,28:21-30);在拟南芥中过表达35s:oswrk45和35s:oswrk72,植物通过诱导aba/胁迫相关基因获得干旱抗性(qiuy.p.,yud.q.,environ.exp.bot.2009,65:35-47);oswrky08的转录可以被peg,nacl或者aba诱导,通过上调非生物胁迫相关基因atcor47和atrd21从而使转基因拟南芥获得对渗透压的抗性(songy.,jings.j.,yud.q.,chin.sci.bull.2009,54:4671-4678);专利cn105441460a公开了一种岷江百合wrky转录因子基因lrwrky1及应用,结果显示超表达lrwrky1的转基因烟草对葡萄座腔菌、核盘菌、灰葡萄孢、尖孢镰刀菌等四种真菌的生长具有明显的抑制作用。但至今有关wrky基因调控植物生长发育的功能研究相对滞后,并且有关wrky基因的研究主要集中于模式植物拟南芥、水稻和杨树上,其它物种中wrky调控因子的研究比较少,研究历史较短,且功能信息缺乏。
岷江百合(liliumregalewilson)是百合科百合属中重要的野生种质资源,广泛分布于我国四川岷江流域(杨利平和符勇耀著,中国百合资源利用研究[m],哈尔滨:东北林业大学出版社,2018.11)。相对于模式植物,岷江百合的生长发育调控机制具有复杂性和特殊性。前人研究表明,岷江百合对百合真菌病和病毒病具有较强的抗性,目前已经从岷江百合中获得多个逆境胁迫相关基因,如lr14-3-3、lrpr10、lrbzip1和lrwrky1等(lih.,etal.,sci.hort.2014,168:9-16;heh.,etal.,genesgenom.2014,36:497-507;zhangn.n.,genesgenom.etal.,2014,36:789-798;hanq.,etal.,sci.hort.2016,198:370-378;专利号201610001896.4),但有关岷江百合wrky转录因子基因调控植物生长发育的研究报道未见报道,其生物学功能及转录调控机制还远未阐明。
技术实现要素:
针对现有技术存在的空白,本发明的目的在于提供一种岷江百合lrwrky2基因,以及使用该基因的cdna序列制备新型的转基因地被植物,填补了百合wrky转录因子基因调控植物生长发育的研究空白。
本发明还提供一种适合庭院和园林绿化中大面积栽培和应用,叶边缘具有规则突起的特殊形态的新产品或方法。
为实现上述目的,本发明采用如下技术方案:一种岷江百合lrwrky2基因,其核苷酸序列如seqidno.1所示。
上述岷江百合lrwrky2基因编码的蛋白,其氨基酸序列如seqidno.2所示。
一种重组载体,包括所述lrwrky2基因的核苷酸序列。
一种遗传工程化的宿主细胞,含有上述重组载体或基因组中整合有lrwrky2基因。
lrwrky2基因或其编码的蛋白可以用于调节拟南芥匍匐生长。
一种匍匐生长拟南芥的制备方法,包括以下步骤:将制备的或提供的含有lrwrky2基因的表达载体转化农杆菌,得到农杆菌工程菌,再将所述农杆菌工程菌浸染拟南芥,使lrwrky2基因过表达,即得到匍匐生长的转基因拟南芥。
优选的,所述转基因拟南芥的茎生叶为莲座叶和/或叶片有规则的叶边缘突起。
相比现有技术,本发明具有如下有益效果:
1、本发明首次克隆了岷江百合lrwrky2基因全长序列,通过氨基酸序列比较分析发现其含有2个典型的wrky功能结构域;进一步构建lrwrky2基因与绿色荧光标记蛋白gfp融合表达载体,转化烟草表皮细胞后分析发现,lrwrky2转录因子具有在细胞核上特异性表达的特征,证实lrwrky2基因属于wrky转录因子家族。
2、本发明首次利用转基因技术调控lrwrky2的cdna序列在拟南芥中过量表达,获得了匍匐生长的转基因模式植物,表明岷江百合lrwrky2基因具有调控草本植物从直立生长到匍匐生长的重要生物学功能。
3、本发明通过在草本模式植物中异源转化百合的一个wrky转录因子基因lrwrky2,不仅彻底改变了植株的生长发育表型,而且调控转基因植株茎上长出多个莲座状的基生叶,单叶边缘具有规则突起的特殊形态,美观新颖,非常适合庭院或绿地的栽培和应用。
4、本发明提供了一种利用岷江百合lrwrky2基因序列制备新型地被植物的方法,对于草本植物的生长发育调控具有重要科学意义,同时获得的转基因地被植物在园艺园林、景观设计领域具有潜在的应用价值。
附图说明
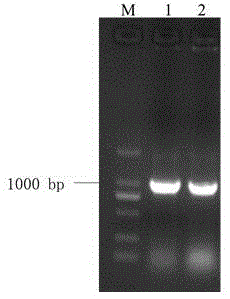
图1为岷江百合lrwrky2基因的pcr扩增电泳图;
图2为构建重组表达载体ptf101-35s::gfp-lrwrky2-nos的图谱示意图;
图3为岷江百合lrwrky2转录因子的亚细胞定位分析;
对照组从左到右依次分别为激发光下暗场图、白光下明场图和组合图;实验组从左到右依次分别是激发光下暗场图、白光下明场图和组合图;
图4为构建重组表达载体pbi121-35s::lrwrky2-nos的图谱示意图;
图5为过表达的转基因拟南芥株系的分子检测示意图;
图a是利用常规pcr从转基因拟南芥的不同株系基因组dna中扩增卡那霉素(kan)标记基因的电泳图;图b是岷江百合lrwrky2基因在过表达转基因株系中的半定量表达分析图;其中m为marker,wt为野生型拟南芥植株,35s::lrwrky2-l1、35s::lrwrky2-l2、35s::lrwrky2-l3分别是转基因拟南芥株系;
图6为过表达lrwrky2的转基因拟南芥植株表型图;
图a是野生型拟南芥植株;图b是过表达lrwrky2的转基因拟南芥植株;
图7为过表达lrwrky2的转基因拟南芥叶片表型图;
图a是野生型拟南芥植株茎上叶片;图b是过表达lrwrky2的转基因拟南芥匍匐茎上早期生长的莲座叶片;图c是转基因拟南芥匍匐茎后期生长的莲座叶片;
图8为过表达lrwrky2的转基因拟南芥的叶边缘图;
从上到下分别是野生型拟南芥的单叶和过表达lrwrky2的转基因拟南芥的单叶;bars代表实际长度1cm。
具体实施方式
下面结合具体实施例和附图对本发明作进一步详细说明。实施例中所述原料如无特殊说明,即为普通市售产品。实施例中所述的实验方法无特别说明,即按常规分子生物学实验方法操作。
实施例1克隆岷江百合lrwrky2全长基因序列
目前,岷江百合(liliumregalewilson)的基因组测序还没有完全启动,课题组前期通过不同组织的转录本测序分析,获得了有关lrwrky2的reads序列,通过序列拼接,初步获得其全长序列;为了验证岷江百合中真实存在的lrwrky2全长基因,进而设计特异性引物(lrwrky2-atg-f和lrwrky2-stop-f)进行pcr扩增分析。
取现蕾期岷江百合叶片组织为材料,采用trizol™plusrnapurificationkit(12183555,invitrogen™),按说明书步骤提取总rna,利用dnasei(18047019,invitrogen™)去除残余的微量dna,并用分光光度计测定rna的浓度,备用。
取约2.0μg岷江百合叶片总rna,按照primescriptiifirst-strandcdnasynthesiskit(6210a,takara)说明书步骤合成第一链cdna。
pcr扩增体系为高保真扩增酶primestarhs(r010a,takara)0.25μl,5×primestarbuffer(mg2+plus)5μl,正向引物(lrwrky2-atg-f,10μm)0.5μl,反向引物(lrwrky2-stop-r,10μm)0.5μl,模板(dna)1μl,dntp(2.5mm)2μl,无菌ddh2o补足至25μl。
正向引物和反向引物的序列如下所示:
lrwrky2-atg-f:tccatggagaacaaaatgggatgg
lrwrky2-stop-r:tctcaccaccacctgaagttgg
pcr反应程序为:预变性95℃,5min;95℃,30s;60℃,40s;72℃,2min,38个循环;72℃,10min。
获得pcr产物通过琼脂糖凝胶电泳分析,如图1所示,在紫外光照射下,在2kb左右可以观察到一条特异性的扩增条带。按照胶回收试剂盒(9672,takara)纯化后备用。
利用平末端加a试剂将纯化的dna片段加a,通过ta克隆将其与pmd20-tvector(6019,takara)相连,连接产物转化大肠杆菌dh5a,从含有氨苄霉素(100mg/l)的lb培养板上挑取2-3个阳性克隆测序进行测序分析,结果表明,岷江百合lrwrky2全长基因序列如seqidno.1所示,含有930bp的开放阅读框(含终止密码)。将lrwrky2全长序列在ncbi数据库进行blast分析,没有发现其同源序列。
实施例2重组表达载体p-tf101-p35s::gfp-lrwrky2-nos构建及亚细胞定位分析
(1)构建p-tf101-p35s::gfp-lrwrky2-nos表达载体
根据岷江百合lrwrky2全长基因序列(seqidno.1),设计引物lrwrky2-gfp-f和lrwrky2-gfp-r,引物中引入无缝克隆(in-fusion)载体接头序列和酶切位点序列。以实施例1中ta连接的阳性克隆质粒为模板,以lrwrky2-gfp-f(正向引物)和lrwrky2-gfp-r(反向引物)为引物进行基因lrwrky2的特异性扩增。
所述引物序列如下:
lrwrky2-gfp-f:
5’-ttccccgggctcgagaagcttatggagaacaaaatgggatgg-3’;
lrwrky2-gfp-r:
5’-ttatctagatccggtggatcctcaccaccacctgaagttgg-3’;
其中,lrwrky2-gfp-f的加粗序列为hindiii酶切位点,lrwrky2-gfp-r的加粗序列为bamhi酶切位点。下划线处为in-fusion克隆载体接头序列。lrwrky2-gfp-f引物序列不含gfp终止密码。
pcr反应体系:高保真扩增酶primestarhs(r010a,takara)0.5μl,5xprimestarbuffer(mg2+plus)10μl,正向引物(10μm)1μl,反向引物(10μm)1μl,模板(50倍稀释的质粒)1μl,dntp(2.5mm)4μl,无菌ddh2o补足至50μl。
pcr反应条件:预变性95℃,5min;95℃,30s;60℃,40s;72℃,2min,38个循环;72℃,7min。
将pcr扩增产物进行琼脂糖凝胶电泳检测(图1)。扩增得到的目的片段与预期片段大小相同,按照胶回收试剂盒(9672,takara)的说明书步骤回收纯化,即获得目的基因片段。
用hindiii和bamhi双酶切处理ptf101-gfp表达载体。酶切体系为:ptf101-gfpvector5μl;hindiii0.5μl;bamhi0.5μl;buffer10xk2μl;无菌ddh2o补足至20μl;37℃反应3h。酶切结束后,根据takara琼脂糖凝胶回收试剂盒回收ptf101-gfp载体片段。
利用无缝克隆技术(in-fusionhdcloningkit,takara)构建p-tf101-p35s::gfp-lrwrky2-nos重组表达载体。
重组反应体系为:
purifedpcrfragment(回收的lrwrky2目的片段)50ng;linearizedvector(ptf101-gfp载体)100ng;5xin-fusionhdenzymepremix2μl;无菌ddh2o补足至10μl。然后按照分子克隆实验指南将上述重组反应体系转化大肠杆菌dh5a,并涂布于含有壮观霉素(spec,100mg/l)的筛选培养板上,通过阳性克隆测序,获得正确的含lrwrky2基因片段的重组表达载体p-tf101-p35s::gfp-lrwrky2-nos(图2)。重组表达载体中报告基因gfp与目的基因lrwrky2的5’端融合后,位于组成型启动子p35s下游,形成融合表达;lrwrky2的3’端组装有nos终止子,可以有效终止融合基因的转录。报告基因gfp经过蓝光激发,无需辅助因子和底物就能发出绿色荧光,作为报告基因可以检测目的基因表达情况。
(2)岷江百合lrwrky2的亚细胞定位分析
1)农杆菌介导的烟草瞬时转化
通过常规冻融法将构建的重组表达载体p-tf101-p35s::gfp-lrwrky2-nos转入农杆菌菌株eha105中,通过pcr筛选阳性克隆。以含有ptf101-gfp质粒的农杆菌菌株为阳性对照,按照霍琳等(农杆菌介导的烟草瞬时表达试验条件优化,分子植物育种,2016,14(1):80-85)方法制备农杆菌注射缓冲液。选择光照培养箱中正常生长具有8-10片叶子完全展开的烟草进行注射,用去除针头的注射器将注射缓冲液缓慢推入叶片背面。然后,把转化后的植株重新放回培养箱中,培养36h-48h后进行观察。
2)gfp报告基因在烟草表皮细胞中的表达与观察
用剪刀小心剪下转化后的烟草叶片放在载玻片上,加1滴蒸馏水,制成装片;然后放在荧光显微镜上,在激发光波长488-507nm的蓝光下,进行荧光观察。结果如图3所示,对照组(图3上)在烟草表皮细胞的细胞核和细胞膜上均有荧光表达,而实验组(图3下)只在烟草表皮细胞的细胞核上特异性表达。以上结果表明,岷江百合lrwrky2是1个核定位的转录因子蛋白,与模式植物中的大部分的wrky因子表达定位情况相符。
实施例3重组表达载体pbi121-p35s::lrwrky2-nos的构建及拟南芥遗传转化
(1)构建pbi121-p35s::lrwrky2-nos过表达载体
根据岷江百合lrwrky2全长基因序列(seqidno.1),设计引物lrwrky2-inf-f和lrwrky2-inf-r,引物中引入无缝克隆(in-fusion)载体接头序列和酶切位点序列。以实施例1中ta连接的阳性克隆质粒为模板,以lrwrky2-inf-f和lrwrky2-inf-r为引物进行基因lrwrky2的特异性扩增。
所述引物序列如下:
lrwrky2-inf-f:
5’-ggactctagaggatccatggagaacaaaatgggatgg-3’
lrwrky2-inf-r:
5’-gatcggggaaattcgagctctcaccaccacctgaagttgg-3’
其中,正向引物lrwrky2-inf-f的加粗序列为bamhi酶切位点,反向引物lrwrky2-inf-r的加粗序列为saci酶切位点。下划线处为in-fusion克隆载体接头序列。
pcr反应体系为:高保真扩增酶primestarhs(r010a,takara)0.5μl,5xprimestarbuffer(mg2+plus)10μl,正向引物(10μm)1μl,反向引物(10μm)1μl,模板(50倍稀释的质粒)1μl,dntp(2.5mm)4μl,无菌ddh2o补足至50μl。
pcr反应条件:预变性95℃,5min;95℃,30s;60℃,40s;72℃,2min,38个循环;72℃,10min。
将pcr扩增产物进行琼脂糖凝胶电泳检测。扩增得到的目的片段与预期片段大小相同,按照胶回收试剂盒(9672,takara)的说明书步骤回收纯化,即获得目的基因片段。
用bamhi和saci双酶切处理pbi121植物双元表达载体。酶切体系为:pbi121vector5μl;bamhi0.5μl;saci0.5μl;buffer10xk1μl;无菌ddh2o补足至20μl;37℃反应3h。酶切结束后,根据takara琼脂糖凝胶回收试剂盒回收pbi121载体大片段。
利用无缝克隆技术(in-fusionhdcloningkit,takara)构建p35s::lrwrky2-nos重组表达载体。
重组反应体系为:
purifedpcrfragment(回收的lrwrky2目的片段)50ng;linearizedvector(pbi121)100ng;5xin-fusionhdenzymepremix2μl;无菌ddh2o补足至10μl。然后按照分子克隆实验指南将上述重组反应体系转化大肠杆菌dh5a,并涂布于含有卡纳霉素(100mg/l)的筛选培养板上,通过阳性克隆筛选和测序,获得正确的含lrwrky2基因片段的重组表达载体pbi121-35s::lrwrky2-nos(图4)。重组表达载体中目的基因lrwrky2的5’端位于组成型启动子p35s下游,它能使lrwrky2基因过量表达;lrwrky2的3’端组装有nos终止子,可以有效终止基因的转录。在重组表达载体上组装有nptii基因,作为转基因植物的筛选标记,可以用卡纳霉素进行转基因植株的筛选。在重组表达载体上组装有lb和rb序列,促使组装于其间的表达框架和筛选标记基因nptii整合至植物受体染色体上。
(2)农杆菌介导的拟南芥遗传转化
拟南芥的遗传转化采用浸花法(zhangx.,etal.,natprotoc.2006,1:641-646)。将携带pbi121-35s::lrwrky2-nos载体的农杆菌导入哥伦比亚col型拟南芥。用卡那霉素(100mg/l)筛选具抗性的拟南芥再生幼苗,通过常规pcr方法扩增抗性标记基因。pcr扩增引物为正向引物(nptii-f:ttgggtggagaggctattcgg)和反向引物(nptii-r:gccacagtcgatgaatccag)。pcr检测结果如图5a所示,在野生型植株(wt)中检测不到nptii标记基因的存在,只有在转基因植株(35s::lrwrky2-l1、35s::lrwrky2-l2和35s::lrwrky2-l3)中扩增nptii标记基因,表明重组表达框已经导入拟南芥基因组。
进一步,通过半定量rt-pcr鉴定得到表现型良好的过表达lrwrky2的转基因株系,采用trizol试剂(invitrogen™),按说明书步骤提取拟南芥叶片总rna,利用dnasei(invitrogen™)去除残余dna,采用cdna反转录试剂(takara)并按照说明书步骤合成第一链cdna。以拟南芥β–actin基因为内参,通过半定量rt-pcr方法分析转基因株系与野生型中lrwrky2基因的表达水平。
检测β–actin基因引物为:
β–actin-f:5'-cacttgcaccaagcagcatgaaga-3'
β–actin-r:5'-aatggaaccaccgatccagacact-3'
检测目的基因引物为:
camv35s-f:agaagacgttccaaccacgtct
lrwrky2-r:tcaccaccacctgaagttgg
结果如图5b所示,在3个代表转基因株系(35s::lrwrky2-l1、35s::lrwrky2-l2和35s::lrwrky2-l3)中目的基因岷江百合lrwrky2均上调表达,并且35s::lrwrky2-l3表达水平为最高,而在野生型植株(wt)中检测不到lrwrky2的表达,表明lrwrky2已经导入拟南芥基因组并成功转录表达。
实施例4转基因拟南芥的表型观察与分析
鉴定了土壤中生长约45天左右的拟南芥植株表型,结果如图6显示,过量表达lrwrky2的转基因植株表现为匍匐生长,而野生型对照植株表现为正常的直立生长。并且过量表达lrwrky2的转基因植株的茎是从基生莲座叶的下部发生,而野生型对照植株的茎是从基生莲座叶的上部发生。最为突出的表型是茎生叶发生明显改变,如图7所示,45天左右野生型对照植株的茎生叶表现为单叶,而转基因植株的茎生叶仍表现为莲座叶,图8显示了60天以后转基因植株茎生叶的莲座叶形态。进一步对转基因莲座叶的观察发现(图8),莲座叶中每个单叶的叶边缘也发生了改变,表现出规则的叶边缘突起。根据前人的报道(bilsboroughg.d.,etal.,pnas2011,108:3424-3429;kasprzewskaa.,etal.,plantj.2015,83:705-718),叶片发生与叶边缘凸起均与生长素信号密切相关,表明岷江百合lrwrky2可能从上游调控了生长素信号途径中的关键调节因子或是直接与之相互作用;进而彻底改变了拟南芥植株的生长发育形态。
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
sequencelisting
<110>长江师范学院;
<120>一种岷江百合lrwrky2基因及应用
<160>14
<170>siposequencelisting1.0
<210>1
<211>930
<212>dna
<213>岷江百合(liliumregalewilson)
<400>1
atggagaacaaaatgggatgggagcagagcttgatatacatgctagctcaagttgaggag60
cagacgaagcaacttgaagcccacctcgacgccgccacaccatcccagatgtgcaagcca120
ctggcacagaagattcgctccaccatatgcacggctatcaccatggtgacggagggttcg180
cagctgcacggcggatcagactcaccgcggtcagccagcgatagcccacggagtgagaac240
tctggcagagctttcacagacgagcaaagggaattgtcgaagaagaggaagaccatgcca300
aaatggagcagccaagtacgggttggttcgggatcagggactgaggcaccaatggatgat360
ggctatagttggaggaagtatgggcagaaggacattctcggagctcaccatccaagggcc420
tactaccggtgcacacatcgcaattctcgtggatgtccggcgacaaagcaagtgcagcga480
tcagacgaggaccctttcgtttttgatgtcatttatagaggtgagcatatatgcgtcgaa540
aatactcagatgattccatgtcaacagaatcagttacagcaaccgccgccacctgcggca600
gctttgcacaccgagttgattccgggtttacagactggtttgcacgttaagacgcaagtt660
atagacttagaagcccaggacccagcgtcgtctttctccttcccatcaacaccgtctagc720
tttgctccggcttctgtctctaacacgtcttccccgacactaggcgggatagctgatact780
tttggtgctggacaatcagaacttgctgagatcttcgctgctgcaactgtggcggaagat840
tcgcctgtagttgacatgaattttgggcttgatcaagtggagctcgacactgcttttcca900
tatgacaattccaacttcaggtggtggtga930
<210>2
<211>309
<212>prt
<213>岷江百合(liliumregalewilson)
<400>2
menkmgweqsliymlaqveeqtkqleahldaatpsqmckplaqkirstictaitmvtegs60
qlhggsdsprsasdsprsensgraftdeqrelskkrktmpkwssqvrvgsgsgteapmdd120
gyswrkygqkdilgahhprayyrcthrnsrgcpatkqvqrsdedpfvfdviyrgehicve180
ntqmipcqqnqlqqppppaaalhtelipglqtglhvktqvidleaqdpassfsfpstpss240
fapasvsntssptlggiadtfgagqselaeifaaatvaedspvvdmnfgldqveldtafp300
ydnsnfrww309
<210>3
<211>24
<212>dna
<213>人工序列
<400>3
tccatggagaacaaaatgggatgg24
<210>4
<211>22
<212>dna
<213>人工序列
<400>4
tctcaccaccacctgaagttgg22
<210>5
<211>42
<212>dna
<213>人工序列
<400>5
ttccccgggctcgagaagcttatggagaacaaaatgggatgg42
<210>6
<211>41
<212>dna
<213>人工序列
<400>6
ttatctagatccggtggatcctcaccaccacctgaagttgg41
<210>7
<211>37
<212>dna
<213>人工序列
<400>7
ggactctagaggatccatggagaacaaaatgggatgg37
<210>8
<211>40
<212>dna
<213>人工序列
<400>8
gatcggggaaattcgagctctcaccaccacctgaagttgg40
<210>9
<211>21
<212>dna
<213>人工序列
<400>9
ttgggtggagaggctattcgg21
<210>10
<211>20
<212>dna
<213>人工序列
<400>10
gccacagtcgatgaatccag20
<210>11
<211>24
<212>dna
<213>人工序列
<400>11
cacttgcaccaagcagcatgaaga24
<210>12
<211>24
<212>dna
<213>人工序列
<400>12
aatggaaccaccgatccagacact24
<210>13
<211>22
<212>dna
<213>人工序列
<400>13
agaagacgttccaaccacgtct22
<210>14
<211>20
<212>dna
<213>人工序列
<400>14
tcaccaccacctgaagttgg20
- 还没有人留言评论。精彩留言会获得点赞!