一种帕博昔布中间体氰基化反应制备杂芳基氰化物的方法与流程
本发明涉及有机合成
技术领域:
,尤其涉及一种帕博昔布中间体氰基化反应制备杂芳基氰化物的方法。
背景技术:
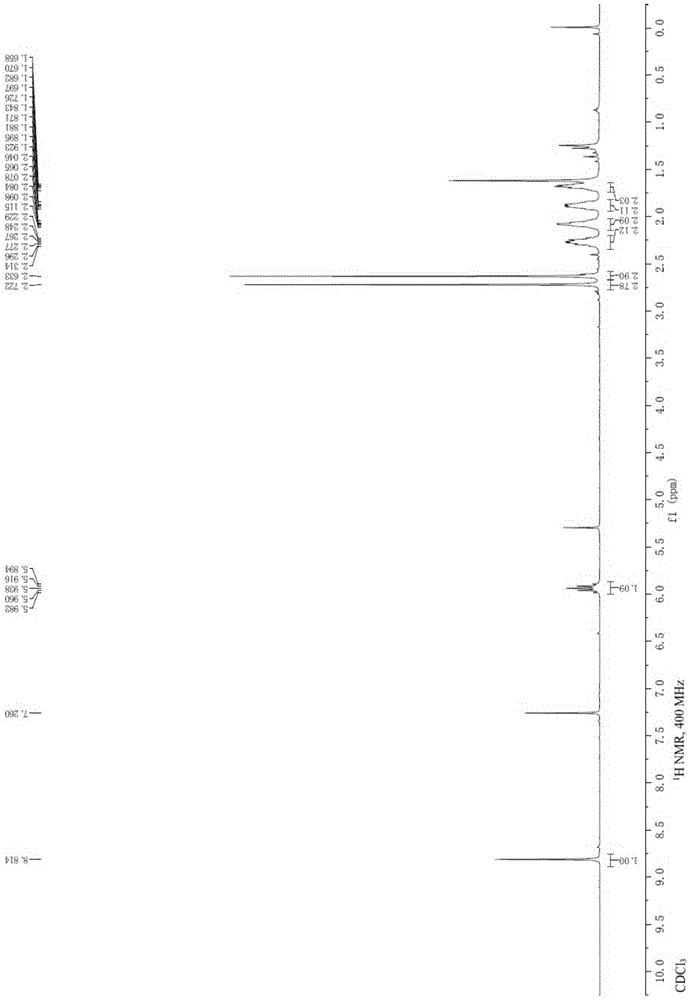
:帕博昔布(palbociclib)是一种高效cdk4/6抑制剂,具有广谱抗癌活性,是全世界在cdk抑制剂领域首个重磅式新药。帕博昔布具有与之前抗肿瘤药机制完全不同的首个cdk4/6的高选择性抑制剂,除了晚期转移性乳腺癌,针对其他多种恶性肿瘤如淋巴癌、肺癌和黑色素瘤等都处在相应的临床实验阶段。在帕博昔布的早期化学合成研究中,主要涉及其核心骨架吡啶并[2,3-d]嘧啶-7-酮化合物的合成及修饰,反应路线i中的化合物2、3和4均为涉及该骨架的重要中间体。化合物2可在六甲基二硅基氨基锂作用下与乙酸乙酯成环得到具有核心骨架吡啶并[2,3-d]嘧啶-7-酮的化合物3,再与n-溴代丁二酰亚胺反应得到6-位溴代物4。帕博昔布的成功将使吡啶并[2,3-d]嘧啶-7-酮类化合物在药物化学研究中得到更多的应用。而此类化合物在6-位溴代后使其在该位置的修饰变化更为丰富,其中最常见的便是溴代位置的氰基化反应。因为芳基氰化物本身就存在于很多染料、除草剂、农用化学品、药物和天然产物中。在药物化学研究中,芳基氰化物更是可以转化为各类有生物活性的重要中间体。由于多种多样的药物分子设计的需要,简单的芳环氰基化方法已不能满足药物合成的需求,类似帕博昔布中间体这样特殊的吡啶并[2,3-d]嘧啶-7-酮类化合物的氮杂芳环氰基化反应需要更多的方法学研究和探索。吡啶并[2,3-d]嘧啶-7-酮是一类高度缺电子的杂环化合物,这类化合物与一般的芳环化合物或富电子的芳环化合物相比,通常的氰基化反应条件都很难实现。现有文献中,仅有少数研究涉及了类似结构的氰基化反应,都用了氰化亚铜(cucn)作为氰基化试剂,反应温度都比较高。部分研究还尝试了用氰化四乙基铵实现氰基化反应,但是该试剂价格昂贵,且产率也不高。中国专利文献上公开了“6-取代的吡啶并[2,3-d]嘧啶类化合物作为蛋白激酶抑制剂”,其申请公布号为cn108191857a,该发明用n-甲基吡咯烷酮(nmp)作为溶剂,在氰化亚铜作用下,能够在150℃的加热条件下以34%的产率得到氰基化产物,但是该方法所需反应温度较高,必需的过量氰化亚铜在高温下吸附大量产物,使得收率降低。上述现有技术虽然能得到氰基化产物,但是产率很难提高,而且所用的氰源,氰化亚铜和氰化四乙基铵,在后处理过程中都会产生对人体和环境有害的氰根离子。所以寻找更为高效的催化体系和更加安全的氰源制备杂芳基氰化物非常重要。技术实现要素:本发明为了克服现有帕博昔布中间体氰基化反应成本高、产率低、会产生对人体和环境有害的氰根离子的问题,提供了一种帕博昔布中间体氰基化反应制备杂芳基氰化物的方法,廉价且绿色环保,反应条件更温和,产率更高,且反应后处理过程不会产生对人体和环境有害的氰根离子,对制药及相关领域的研究和工业化发展具有重要意义。为了实现上述目的,本发明采用以下技术方案:一种帕博昔布中间体氰基化反应制备杂芳基氰化物的方法,按照以下化学反应方程式进行:在惰性气体保护下,将6-卤代吡啶并[2,3-d]嘧啶-7-酮类化合物(i),氰基化试剂,催化剂,醋酸钾,n,n-二甲基甲酰胺和水混合于反应瓶中,密封后加热搅拌至反应完全;反应液冷却后加入乙酸乙酯,然后用饱和食盐水洗涤,所得有机相经无水硫酸钠干燥后过滤并浓缩,粗品经柱层析后得到6-氰基吡啶并[2,3-d]嘧啶-7-酮类化合物(ii)。本发明解决了目前药物合成及其它化学相关领域中,对于具有吡啶并[2,3-d]嘧啶-7-酮骨架的氮杂环类化合物的6-位氰基化反应的问题,且对其它尚未被研究的具有缺电子芳环骨架的化合物的氰基化反应有启示性作用。由于各类芳基或杂芳基氰化物在药物化学研究中是转化为各种活性分子的重要中间体,本发明大量扩充了可合成的氰化物的范围。与现有技术相比,反应条件更温和,产率更高,且反应后处理过程不会产生对人体和环境有害的氰根离子,对制药及相关领域的研究和工业化发展具有重要意义。作为优选,所述6-卤代吡啶并[2,3-d]嘧啶-7-酮类化合物具有以下化学式:其中,r1为甲硫基,氨基或烃基、芳基、杂环基、芳杂环基取代的氨基;r2为h或c1~c10的烷基;r3为c1~c10的烷基或c3~c6的环烷基;x为溴原子,氯原子或碘原子。作为优选,所述氰基化试剂为六氰基亚铁酸钾,其化学分子式为k4[fe(cn)6],通常以三水合物(k4[fe(cn)6]·3h2o)的形式存在,也被叫做黄血盐,是一种对人体和环境无害的,非常安全的氰基化试剂,其甚至可以作为抗凝结剂被添加于食用盐中。无论这种试剂本身还是其参与反应后形成的副产物,都不会对人体或环境产生危害。本发明仅使用廉价且对环境无毒害的六氰基亚铁酸钾作为氰源即可实现高效的氰基化反应,是适合工业放大使用的理想氰基化反应试剂。作为优选,所述催化剂为双[2-(二苯基膦基)苯基]醚氯化钯(pdcl2(dpephos)),其化学分子式为c36h28cl2op2pd,是一种已经商品化的双膦配体钯催化剂,其结构式为:双[2-(二苯基膦基)苯基]醚(dpephos)与氯化钯形成的络合物催化剂pdcl2(dpephos)能够有效优化本发明的氰基化反应,因为双[2-(二苯基膦基)苯基]醚与氯化钯特殊的宽角度双膦配位,对缺电子的芳环化合物是一种很好的催化剂体系。因此,本发明具有如下有益效果:原料成本低,绿色环保,反应条件更温和,产率更高,且反应后处理过程不会产生对人体和环境有害的氰根离子,对制药及相关领域的研究和工业化发展具有重要意义。附图说明图1是实施例1制得的产物的核磁共振1h谱图。图2是实施例2制得的产物的核磁共振1h谱图。具体实施方式下面通过具体实施例,并结合附图,对本发明的技术方案作进一步具体的说明。在本发明中,若非特指,所有设备和原料均可从市场购得或是本行业常用的,下述实施例中的方法,如无特别说明,均为本领域常规方法。实施例1按照以下化学反应方程式,6-溴-8-环戊基-5-甲基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮氰基化反应制备6-氰基-8-环戊基-5-甲基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮:在惰性气体保护下,向25ml反应瓶中加入6-溴-8-环戊基-5-甲基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮(4,500mg,1.41mmol),六氰基亚铁酸钾三水合物(298mg,0.71mmol),双[2-(二苯基膦基)苯基]醚氯化钯(303mg,0.42mmol),醋酸钾(69.3mg,0.71mmol),n,n-二甲基甲酰胺(5ml)和水(0.5ml),反应瓶用橡皮塞密封后在100℃加热并搅拌7小时。反应液冷却后加入乙酸乙酯,然后用饱和食盐水洗涤,所得有机相经无水硫酸钠干燥后过滤并浓缩,粗品经柱层析后得到的浅黄色固体即为6-氰基-8-环戊基-5-甲基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮(5,342mg,产率:80.7%)。本实施例制得的产物的核磁共振1h谱图如图1所示:1hnmr(400mhz,cdcl3,ppm):δ8.81(s,1h),5.98–5.89(m,1h),2.72(s,3h),2.63(s,3h),2.31–2.23(m,2h),2.12–2.05(m,2h),1.92–1.84(m,2h),1.73–1.66(m,2h).实施例2按照以下化学反应方程式,6-溴-8-环戊基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮氰基化反应制备6-氰基-8-环戊基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮:在惰性气体保护下,向25ml反应瓶中加入6-溴-8-环戊基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮(4a,680.5mg,2mmol),六氰基亚铁酸钾三水合物(422.4mg,1mmol),双[2-(二苯基膦基)苯基]醚氯化钯(429.5mg,0.6mmol),醋酸钾(98.1mg,1mmol),n,n-二甲基甲酰胺(7ml)和水(0.7ml),反应瓶用橡皮塞密封后在100℃加热并搅拌7小时。反应液冷却后加入乙酸乙酯,然后用饱和食盐水洗涤,所得有机相经无水硫酸钠干燥后过滤并浓缩,粗品经柱层析后得到的浅黄色固体即为6-氰基-8-环戊基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮(5a,494.2mg,产率:86.3%)。本实施例制得的产物的核磁共振1h谱图如图2所示:1hnmr(400mhz,cdcl3,ppm):δ8.67(s,1h),8.09(s,1h),5.99–5.90(m,1h),2.64(s,3h),2.33–2.24(m,2h),2.11–2.04(m,2h),1.95–1.86(m,2h),1.74–1.67(m,2h).实施例3按照以下化学反应方程式,4-(6-(6-溴-8-环戊基-5-甲基-7-氧代-7,8-二氢吡啶并[2,3-d]嘧啶-2-基氨基)吡啶-3-基)哌嗪-1-甲酸叔丁酯氰基化反应制备4-(6-(6-氰基-8-环戊基-5-甲基-7-氧代-7,8-二氢吡啶并[2,3-d]嘧啶-2-基氨基)吡啶-3-基)哌嗪-1-甲酸叔丁酯:在惰性气体保护下,向25ml反应瓶中加入4-(6-(6-溴-8-环戊基-5-甲基-7-氧代-7,8-二氢吡啶并[2,3-d]嘧啶-2-基氨基)吡啶-3-基)哌嗪-1-甲酸叔丁酯(4b,584.5mg,1mmol),六氰基亚铁酸钾三水合物(211.2mg,0.5mmol),双[2-(二苯基膦基)苯基]醚氯化钯(214.8mg,0.3mmol),醋酸钾(49.1mg,0.5mmol),n,n-二甲基甲酰胺(4ml)和水(0.4ml),反应瓶用橡皮塞密封后在100℃加热并搅拌7小时。反应液冷却后加入乙酸乙酯,然后用饱和食盐水洗涤,所得有机相经无水硫酸钠干燥后过滤并浓缩,粗品经柱层析后得到的浅黄色固体即为6-氰基-8-环戊基-2-甲硫基吡啶并[2,3-d]嘧啶-7-酮(5b,348mg,产率:65.6%)。对比例1按照以下化学反应方程式:选用氰化锌(zn(cn)2)作为氰基化试剂,在三(二亚苄基丙酮)二钯(pd2(dba)3)催化作用下,以1,1'-双(二苯基膦基)二茂铁(dppf)为配体,进行氰基化反应。对比例2对比例2与对比例1的区别在于:反应体系中加入活化过的锌粉(zn),其余工艺完全相同。对比例3对比例3与对比例1的区别在于:反应体系中加入活化过的锌粉(zn)和醋酸锌(zn(oac)2)作为共同催化体系,其余工艺完全相同。对比例1-3的反应体系及结果如表1所示:表1.对比例1-3得反应体系及结果编号附加反应试剂结果对比例1/原料不反应对比例2zn产率:8.6%,原料4回收60.7%对比例3zn,zn(oac)2产率:11.8%,原料4回收54%为了避免使用氰化钠(nacn)、氰化钾(kcn)这类毒性高,容易造成环境污染的氰基化试剂,本发明申请人首先选用氰化锌(zn(cn)2)来尝试该反应,结果如表1所示,在三(二亚苄基丙酮)二钯(pd2(dba)3)催化作用下,以1,1'-双(二苯基膦基)二茂铁(dppf)为配体,使用氰化锌能够对吡啶类氮杂环卤化物进行高效的氰基化反应。然而,如对比例1所示,该条件在本发明的吡啶并[2,3-d]嘧啶-7-酮骨架中则不能反应。对比例2加入活化过的锌粉(zn),希望能够避免由氰根离子造成的反应失活问题,而且锌粉能够去除反应体系中的氧气,使钯催化剂保持在零价的高效催化状态,此条件能够使反应进行,但是只能以8.6%的产率得到产物,且依然有60.7%的原料不能被转化。对比例3采用锌粉和醋酸锌(zn(oac)2)的共同催化体系,因为醋酸锌能够使催化剂保持更加长久的活度直至反应完全。不过这样的催化体系对本发明研究的底物提高甚微,仅以11.8%的产率得到氰基化产物,且有54%原料被回收。对比例4按照以下化学反应方程式:选用氰化亚铜(cucn)作为氰基化试剂,n-甲基吡咯烷酮(nmp)作为溶剂,反应温度210℃,进行氰基化反应。对比例5对比例5与对比例4的区别在于:反应温度150℃,其余工艺完全相同。对比例6对比例6与对比例4的区别在于:反应温度120℃,其余工艺完全相同。对比例7对比例7与对比例4的区别在于:加入四(三苯基膦)钯作为催化剂,n,n-二甲基甲酰胺(dmf)作为溶剂,反应温度135℃,其余工艺完全相同。对比例4-7的反应体系及结果如表2所示:表2.对比例4-7的反应体系及结果编号催化剂溶剂温度转化率a产率对比例4/nmp210℃100%25.9%对比例5/nmp150℃100%34.2%对比例6/nmp120℃~50%/对比例7pd(pph3)4dmf135℃100%38.2%本发明申请人采用了比较传统的氰源,氰化亚铜(cucn)来尝试该氰基化反应,并做了更为细化的研究。由于此方法需要温度较高,所以选用沸点较高的n-甲基吡咯烷酮(nmp)作为溶剂,发现反应在高温下能够将原料完全转化,但是由于该条件必需的过量氰化亚铜在高温下易吸附产品,并且产生较多杂质,所以对比例4在210℃时产率仅为25.9%;对比例5温度降到150℃时产率能提高到34.2%;对比例6在120℃时近一半原料未反应完,产品很难经纯化得到。对比例7加入四(三苯基膦)钯作为催化剂,能在较低温度下使反应完全转化,产率也略微提高到了38.2%,而溶剂则用沸点相对较低的n,n-二甲基甲酰胺(dmf)即可。对比例1-3和对比例4-7的反应方法虽然都能得到氰基化产物5,但是产率都很难再得到提高,而且所用的氰源,氰化锌和氰化亚铜,在后处理过程中都会产生对人体和环境有害的氰根离子。所以寻找更为高效的催化体系和更加安全的氰源非常重要。对比例8-12对比例8-12与实施例1的区别在于:pdcl2(dpephos)催化剂当量,dmf:h2o溶剂体积比及反应温度不同,具体参见表3,其余工艺完全相同。表3.对比例7-11与实施例2得反应体系及产率a薄层层析色谱(tlc)显色的估计值;b根据回收得到的溴代物原料4推算。将上述对比例及实施例的转化率及产率记录于表3,分析数据可得:对比例8使用0.2当量(底物加料量的20mol%)的催化剂pdcl2(dpephos),在n,n-二甲基甲酰胺(dmf)和水(体积比dmf:h2o=2:1)的混合溶剂体系中,仅用0.5当量(底物加料量的50mol%)的六氰基亚铁酸钾在130℃进行反应,发现原料很快能够反应完,但可能由于温度过高,产生杂质过多,很难分离纯化得到产品。对比例9反应温度降到90℃,反应体系虽然干净了许多,但是有近一半原料不能反应完全,依然没有办法分离到较纯的产品。对比例10将反应温度控制在100℃,发现该温度条件能够提升转化率的同时,还能顺利的分离到34.2%的产品。但是这个条件下仍然有30%没有转化的原料被回收了,我们随后在重复此条件时发现反应体系中始终有颜色类似催化剂的不溶物悬浮在反应液中。对比例11将溶剂比例调节至dmf:h2o=10:1,转化率有了大幅度提高,也能以64.1%的收率得到氰基化产物5。实施例2将催化剂pdcl2(dpephos)的当量提高到0.3时,则实现了100%的转化率和80.7%的分离产率。对比例12只使用dmf的单溶剂体系,转化率则反而降低。以上所述仅为本发明的较佳实施例,并非对本发明作任何形式上的限制,在不超出权利要求所记载的技术方案的前提下还有其它的变体及改型。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1