一种从头孢菌素C发酵液中分离纯化脱乙酰氧基头孢菌素C的方法与流程
本发明涉及分离纯化领域,尤其涉及一种从头孢菌素c发酵液中分离纯化脱乙酰氧基头孢菌素c的方法。
背景技术:
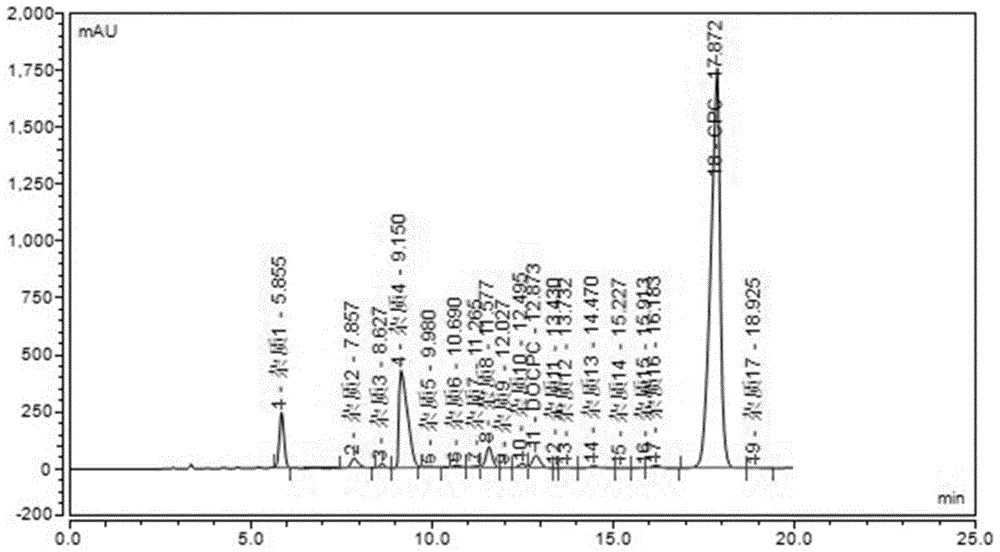
:头孢菌素属β内酰胺类抗生素家族成员,目前占我国抗生素类药物市场份额高达51%,青霉素约占17%。头孢菌素c(cpc)生物合成类似于青霉素,为确定发酵终点、合理补料以便控制发酵过程,必须有效地检测发酵过程中培养基成份、早期杂质及发酵产物与副产物比例。头孢菌素c发酵过程中包括有cpc、脱乙酰头孢菌素c(dcpc)、脱乙酰氧基头孢菌素c(docpc)等物质,脱乙酰氧基头孢菌素c(docpc)是头孢菌素c(cpc)发酵过程中的副产物,为头孢菌素c的结构类似物。由于脱乙酰氧基头孢菌素c可通过结构修饰转化为某些重要的新型头孢类药物的中间体,引起人们的广泛关注。目前尚未有对docpc进行纯化分离的相关报道,如何从头孢菌素c发酵液中分离纯化得到高纯度的docpc具有一定难度且具有很重要的现实意义。技术实现要素:本发明的目的在于提供一种从头孢菌素c发酵液中分离纯化脱乙酰氧基头孢菌素c的方法。本发明所采取的技术方案是:本发明的目的之一在于提供一种从头孢菌素c发酵液中分离纯化脱乙酰氧基头孢菌素c的方法,包括如下步骤:1)取头孢菌素c发酵液进行浓缩,得头孢菌素c浓缩液;2)将头孢菌素c浓缩液进行液相色谱制备柱分离,得脱乙酰氧基头孢菌素c。优选地,步骤1)浓缩至头孢菌素c浓缩液的体积为头孢菌素c发酵液体积的1/6~1/4。优选地,步骤2)的液相色谱制备柱处理,包括如下步骤:ⅰ)将头孢菌素c浓缩液进行第一次液相色谱制备柱处理,得溶液β;ⅱ)将溶液β进行第二次液相色谱制备柱处理,得脱乙酰氧基头孢菌素c。优选地,步骤ⅰ)采用c18填料色谱制备柱进行处理;优选地,制备柱填料为peaceods。优选地,步骤ⅰ)利用流动相a为乙腈,流动相b为体积分数为0.1%的甲酸水溶液进行梯度洗脱。优选地,步骤ⅰ)梯度洗脱中洗脱时间和流动相的体积比为:0~60min1%~12%流动相a。优选地,步骤ⅰ)上述色谱制备柱中的流速为10~12ml/min;更优选为10ml/min。优选地,步骤ⅰ)上述色谱制备柱中的柱温为20~25℃。优选地,步骤ⅰ)中的检测波长为254nm。优选地,步骤ⅱ)采用sepaxgp-c18或welchxtimatec18色谱制备柱进行处理。优选地,步骤ⅱ)利用流动相c为体积比为0.1:100的甲酸乙腈混合溶液,流动相d为体积分数为0.1%的甲酸水溶液进行梯度洗脱。优选地,步骤ⅱ)梯度洗脱中洗脱时间和流动相的体积比为:0~40min3.5%流动相c。优选地,步骤ⅱ)色谱制备柱中的流速为3.0~3.5ml/min。优选地,步骤ⅱ)色谱制备柱中的柱温为30℃。优选地,步骤ⅱ)中的检测波长为254nm。本发明的有益效果是:本发明首次从头孢菌素c发酵液中分离出docpc,并且本发明分离纯化得到的docpc的纯度在97%以上,这说明本发明能够有效地从头孢菌素c发酵液中分离纯化得到高纯度的docpc,这不仅为docpc的药理研究提供了可靠的基础,同时也为进一步得到高纯度的docpc奠定了基础,具有非常重要的现实意义。附图说明图1为实施例1头孢菌素c浓缩液的高效液相色谱图;图2为实施例1溶液β的高效液相色谱图;图3为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的高效液相色谱图;图4为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的核磁共振氢谱图;图5为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的核磁共振碳谱图;图6为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的红外光谱图;图7为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的紫外光谱图;图8为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的质谱图;图9为对比例1分离纯化得到的脱乙酰氧基头孢菌素c的制备液相色谱图;图10为实施例1分离纯化得到的脱乙酰氧基头孢菌素c的制备液相色谱图。具体实施方式下面进一步列举实施例以详细说明本发明。同样应理解,以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明阐述的原理做出的一些非本质的改进和调整均属于本发明的保护范围。下述示例具体的工艺参数等也仅是合适范围中的一个示例,即本领域技术人员可以通过本文的说明做合适范围内的选择,而并非要限定于下文示例的具体数据。下述实施例中的头孢菌素c发酵液由焦作健康元生物制品有限公司提供。实施例1一种从头孢菌素c发酵液中分离纯化脱乙酰氧基头孢菌素c的方法,包括如下步骤:1)取一定量的头孢菌素c发酵液,经旋转蒸发仪(水浴温度40℃)减压浓缩至原体积的1/6~1/4,得头孢菌素c浓缩液;2)将头孢菌素c浓缩液进行高压液相色谱制备柱处理,具体包括如下步骤:ⅰ)将头孢菌素c浓缩液进行第一次高压液相色谱制备柱处理,高压液相色谱柱处理条件为:采用c18填料色谱制备柱,填料为peaceods,流动相a为乙腈,流动相b为体积分数为0.1%的甲酸水溶液进行梯度洗脱,梯度洗脱的洗脱时间和流动相的体积比为:0~60min1%~12%流动相a,色谱制备柱中的流速为10ml/min,色谱制备柱的柱温为20~25℃,检测波长为254nm,得溶液β;ⅱ)将溶液β进行第二次高压液相色谱制备柱处理,高压液相色谱柱处理条件为:采用色谱制备柱为sepaxgp-c18,流动相c为体积比为0.1:100的甲酸-乙腈溶液,流动相d为体积分数为0.1%的甲酸水溶液进行梯度洗脱,梯度洗脱中的洗脱时间和流动相的体积比为:0~40min3.5%流动相c,色谱制备柱中的流速为3.5ml/min,色谱制备柱的柱温为30℃,检测波长为254nm,后经旋转蒸发仪(水浴温度40℃)浓缩,再将浓缩液经冻干机冻干后得脱乙酰氧基头孢菌素c。检测分析:将实施例1中步骤1)制备得到的头孢菌素c浓缩液进行高效液相色谱分析,检测条件如下:色谱柱为zorbaxsb-aq,4.6×250mm,5μm,流动相a为水-0.1%fa,流动相b为乙腈-0.1%fa进行梯度洗脱,梯度洗脱中洗脱时间和流动相a的体积比的顺序为:0~20min:99%~95.5%,20~30min:95.5%~90%,30~40min:90%~80%,色谱柱中的流速为1.0ml/min,色谱柱的柱温为30℃,检测波长为254nm,色谱图见图1,数据见下表1。表1峰编号峰名称保留时间(min)峰面积峰高相对峰面积%1杂质15.85535.946242.6414.692杂质27.85711.68944.1661.533杂质38.6273.69921.3310.484杂质49.15117.115429.88515.295杂质59.983.9237.4830.516杂质610.694.29813.5460.567杂质711.2653.1479.9080.418杂质811.57720.76195.362.719杂质912.0271.1094.2670.1410杂质1012.4955.48621.2730.7211docpc12.87314.98157.3051.9612杂质1113.430.1841.0750.0213杂质1213.7320.6631.7650.0914杂质1314.473.93211.550.5115杂质1415.2270.2590.7790.0316杂质1515.9130.7853.7080.117杂质1616.1834.23312.7310.5518cpc17.872532.9881755.52169.5719杂质1718.9250.9682.4820.13由图1和表1可知:头孢菌素c浓缩液中含有docpc,同时含有较多的杂质,且docpc周围存在与其保留时间相差不大的其他杂质,因此存在非常大的分离难度。将经步骤ⅰ)处理得到的溶液β按照上述高效液相色谱分析方法进行分析,结果见图2及表2:表2峰名称保留时间(min)峰面积峰高相对峰面积(%)19.6780.21790.851.132(杂质7)11.23811.499846.1159.573(杂质9)12.2022.16948.6511.244(docpc)12.8285.416919.8828.06由图2和表2可知:经步骤ⅰ)处理后,溶液β中只剩3个杂质,并且docpc的相对峰面积(即docpc的含量)也有相依提高,这说明经步骤ⅰ)能有效去除大部分杂质。将经步骤ⅱ)处理得到的脱乙酰氧基头孢菌素c溶于水得脱乙酰氧基头孢菌素c水溶液并按上述高效液相色谱分析方法进行分析,结果见图3;由图3可知:实施例1制备得到的脱乙酰氧基头孢菌素c和原头孢菌素c浓缩液中的脱乙酰氧基头孢菌素c的保留时间基本一致,这说明,本发明成功分离得到脱乙酰氧基头孢菌素c,同时采用峰面积归一化方法计算得到实施例1得到的脱乙酰氧基头孢菌素c的纯度为97%;进一步地,将实施例1制备得到的脱乙酰氧基头孢菌素c进行核磁共振(氢谱、碳谱)、红外光谱、紫外光谱及质谱分析,结果见图4~8,氢谱、碳谱数据详见下表3;表3注:表3中的“-”表示没有该数据。由图4~8、表3进一步分析得到本发明分离纯化得到的结构式如下,该结构即为脱乙酰氧基头孢菌素c,这进一步说明了本发明成功分离纯化得到脱乙酰氧基头孢菌素c:对比例1对比例1的分离纯化步骤同实施例1,不同之处在于步骤ⅱ)中的梯度洗脱中的洗脱时间和流动相的体积比为:0~40min4.5%流动相c,也就是说步骤ⅱ)的具体步骤如下:步骤ⅱ)将溶液β进行第二次高压液相色谱制备柱处理,高压液相色谱柱处理条件为:采用色谱制备柱为sepaxgp-c18,流动相c为体积比为0.1:100的甲酸-乙腈溶液,流动相d为体积分数为0.1%的甲酸水溶液进行梯度洗脱,梯度洗脱中的洗脱时间和流动相的体积比的顺序为:0~40min4.5%流动相c,色谱制备柱中的流速为3.5ml/min,色谱制备柱的柱温为30℃,检测波长为254nm,后经旋转蒸发仪(水浴温度40℃)浓缩,再将浓缩液经冻干机冻干后得脱乙酰氧基头孢菌素c。经对比例1和实施例1不同的步骤ⅱ)处理得到的制备液相色谱图见图9和图10,由图9和图10可知:图9的主峰(即:docpc)和杂质的保留时间远比图10的接近,这说明经对比例1处理后其无法将docpc与杂质进行有效分离,需要后续再继续分离纯化才能得到纯度较高的docpc,而实施例1则无需再进行分离纯化即可获得纯度较高的docpc。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1