一种吡唑解草酯杂质、制备方法与应用与流程
本发明涉及药物合成技术领域,具体涉及一种吡唑解草酯杂质、制备方法与应用,该杂质的化学名称为1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯。
背景技术:
吡唑解草酯(mefenpyr-diethyl)是1999年英国brighton植保会议公布的新的除草剂安全剂,是安万特公司开发的吡唑类解毒剂,与除草剂如甲基二磺一起使用可使小麦、大麦等免受伤害。但吡唑解草酯的合成过程中难免会产生一些杂质,这些杂质不仅直接关乎吡唑解草酯的使用效果,还会环境安全和农产品安全产生重要影响,从而对人体健康存在安全隐患,同时这些杂质也成了判定吡唑解草酯产品质量和使用安全性的重要指标之一。但由于吡唑解草酯中杂质的种类较多且杂质的总体含量较少,一直没有被人们成功分离和确认物质信息。本发明目的在于,提出一种吡唑解草酯中含量较高的杂质分子结构信息、制备方法与应用,该杂质的化学名称为1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯。
技术实现要素:
本发明的目的在于,克服现有技术中存在的缺陷,首先,提供了吡唑解草酯杂质中含量较高的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的结构信息;其次,提供一种1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的制备方法,该制备方法具有反应条件温和、耗能少、经济可行高的优点,其产率达40%以上;第三,1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯作为杂质对吡唑解草酯药效的影响评估以及其在农产品上残留的安全评估。
一种吡唑解草酯杂质,其化学名称为1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯,其分子结构式为式ⅰ:
为了便于对1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯进行研究,现提出一种1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的制备方法,包括如下步骤:
s1:制备2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯,以2,4,-二氯苯胺、亚硝酸盐和2-氯乙酰乙酸乙酯为原料,经过重氮化和雅普-克林格曼反应,得2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯,反应式为:
s2:制备1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯,以步骤s1制备的2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯和甲基丙烯酸乙酯为原料,经过环加成反应,得1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯,反应式为:
优选的技术方案是,所述步骤s1的具体操作为:将2,4,-二氯苯胺溶解于乙醇和水的混合溶剂中,加入一定量的浓盐酸,并控制温度为0℃,滴加亚硝酸盐水溶液;然后,加入2-氯乙酰乙酸乙酯,在0℃条件下,反应4~5h,升温至室温,将反应产物混合液依次经过萃取、洗涤、干燥和柱层析,得2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯,其中2,4,-二氯苯胺∶亚硝酸盐∶2-氯乙酰乙酸乙酯∶加入浓盐酸中氯化氢分子的摩尔比为1∶1.1∶1∶2.5。
进一步优选的技术方案还有,所述步骤s1中的亚硝酸盐为亚硝酸钠或亚硝酸钾。
优选的技术方案还有,所述步骤s2的具体操作为:将2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯溶于四氢呋喃中,加入一定量的对甲苯磺酸,然后滴加甲基丙烯酸乙酯,滴加完毕后升温至80℃,反应10~12h,冷却至室温,将反应产物混合液依次经过萃取、洗涤、浓缩和柱层析,得1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯,其中2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯∶对甲苯磺酸∶甲基丙烯酸乙酯的摩尔比为1∶1.5∶3。
为了便于对1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯进行晶体结构分析,现提出一种1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯晶体的培养方法,将步骤s2制备的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯溶于甲醇溶剂中,室温条件下,静置3~5天,析出晶体。
为了便于上述1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的推广应用,现提出一种1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的用途:作为杂质对吡唑解草酯药效的影响评估以及其在农产品上残留的安全评估。
本发明的优点和有益效果在于:
本发明提出了一种在吡唑解草酯杂质中含量较高的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的结构信息及其制备方法,其制备方法为:以2,4,-二氯苯胺、亚硝酸盐和2-氯乙酰乙酸乙酯为起始原料,经过重氮化和雅普-克林格曼反应后得到中间体2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯;然后再以四氢呋喃为溶剂,中间体2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯与甲基丙烯酸乙酯发生闭环反应得到反应产物混合液,反应产物混合液经萃取,洗涤、浓缩和柱层析等处理工序,最终得1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯,收率达到40%以上;且该方法具有反应条件温和、耗能少、经济可行高、产率较高的优点;1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯作为杂质用于对吡唑解草酯药效的影响评估以及其在农产品上残留的安全评估。
附图说明
图1为实施例1中1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的分子结构式;
图2为实施例1中1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的1hnmr图谱;
图3为实施例1中1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的ms图谱;
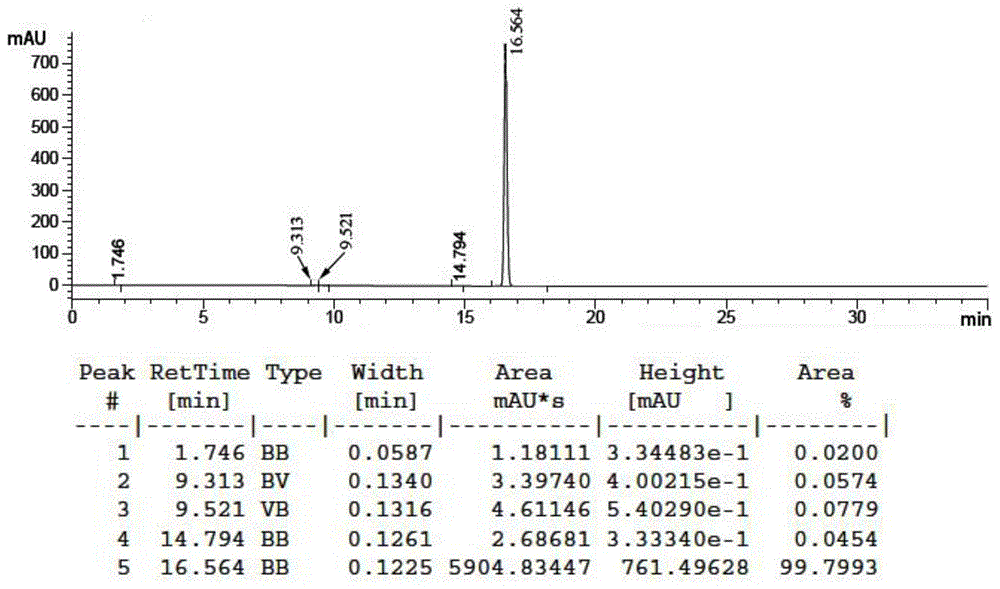
图4为实施例1中1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的高效液相色谱图;
图5为实施例1中1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的晶体结构图。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
实施例1
采用本发明制备方法制备1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯,包括如下步骤:
s1:取一250ml三口烧瓶,加入2,4,-二氯苯胺(10.2g,63.4mmol),加入乙醇(20ml)、水(7ml)和浓盐酸(13ml),控制温度为0℃,在搅拌状态下向三口烧瓶内滴加亚硝酸钠水溶液(nano2:4.69g,69.7mmol;h2o:15ml),滴加完毕继续反应30分钟,然后向三口烧瓶内加入2-氯乙酰乙酸乙酯(10.88g,63.4mmol)、醋酸钠(15.6g,190.1mmol)和水(90ml),继续在0℃条件下搅拌反应4~5h,并用薄层色谱法(thinlayerchromatography,tlc)检测反应进程,tlc所用的展开剂为石油醚和乙酸乙酯混合溶液(二者体积比为5:1),反应完成后升至室温,并将反应产物混合液依次经萃取、洗涤、干燥和柱层析分离纯化,得中间体2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯(14.3g),中间体收率为76%。
s2:将2-氯-2-(2,4-二氯苯腙基)-乙醛酸乙酯(5.9g,0.02mol)溶解于thf(50ml)中,室温搅拌条件下,加入对甲苯磺酸(5.2g,0.03mol),然后滴加甲基丙烯酸乙酯(6.8g,0.06mol),滴加完毕升温至80℃,80℃条件下搅拌反应12小时,tlc检测反应进程,tlc所用的展开剂为石油醚和乙酸乙酯混合溶液(二者体积比为3:1),反应完成后冷却至室温,并将反应产物混合液依次经萃取、洗涤、干燥和柱层析分离纯化,得1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯白色固体(5.3g),收率为42%,纯度为99.8%。
s3:将步骤s2制备的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯(100mg)溶解于5ml甲醇溶剂中,室温条件下,静置3~5天,自然挥发溶剂析出晶体。
1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的分子结构式见附图1;
用buukerav-400型核磁共振仪对步骤s2制备的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯白色固体进行核磁共振分析,结果见附图2;
用thermoltq-orbitrapxl质谱仪对步骤s2制备的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯白色固体进行质谱分析,结果见附图3;
用高效液相色谱仪对步骤s2制备的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯白色固体进行纯度分析,结果见附图4;
用brukersmartapexccd单晶衍射仪对步骤s3培养的1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯单晶进行结构分析,选取大小为0.11mm×0.15mm×0.9mm的无色晶体进行晶体结构测定,采用brukersmartapexccd单晶衍射仪,使用经过石墨单色器单色化的mokα射线(λ=0.71073nm),以φ~ω扫描方式收集衍射数,结果见附图5。
附图2核磁氢谱数据为1h-nmr:δppm7.729to7.734(d,1h),7.516to7.544(m,2h),7.402to7.448(m,2h),7.335to7.357(d,1h),4.260to4.400(m,2h),4.096to4.148(m,2h),3.960to3.997(d,1h),3.684to3.762(m,2h),3.652to3.689(d,1h),1.205to1.240(t,3h),0.952to0.987(t,3h),0.830to0.865(t,3h),与1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯分子式c26h26cl4n4o6中氢原子出峰位置一致;
附图3质谱数据表明,用质谱仪(thermoltq-orbitrapxl),溶剂甲醇,positivepolarityesi(+)/esi,质谱esi(+)结果[m+h]+的m/z为631.2,[(m-ch3ch2o+h)+h]+的m/z为587.1,[(m-ch3ch2o+h-ch3ch2+h)+h]+的m/z为559.0,可以推测样品分子量为630.1,与c26h26cl4n4o6理论分子量相符;
附图4,液相色谱图显示:产物的归一含量为99.8%,纯度较高;
附图5,经过shelxtl作图软件解出化合物的晶体结构,从晶体结构图5上可以看出晶体结构与1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯化合物的分子结构式一致。
为了便于上述1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的推广应用,现提出一种1,5-二(2,4-二氯苯基)-6-甲基-1h,5h-吡唑并[5,1-c]-1,2,4-三唑-3,6,7a-三羧酸三乙酯的用途:作为杂质对吡唑解草酯药效的影响评估以及其在农产品上残留的安全评估。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!