一种他达拉非的制备方法与流程
本发明属于化学合成技术领域。更具体地,涉及一种他达拉非的制备方法。
背景技术:
他达拉非是环磷酸鸟苷(cgmp)特异性磷酸二酯酶5(pde5)的选择性、可逆性抑制剂,经fda批准,于2003年作为治疗男性勃起功能障碍(med)的药物在美国上市。近期临床研究发现,他达拉非还可以用来治疗肺动脉高压,应用前景广泛。与类似药西地那非或伐地那非相比,他达拉非具有活性高、起效快、药效长、副作用小等优势,一直是研究开发的热点。
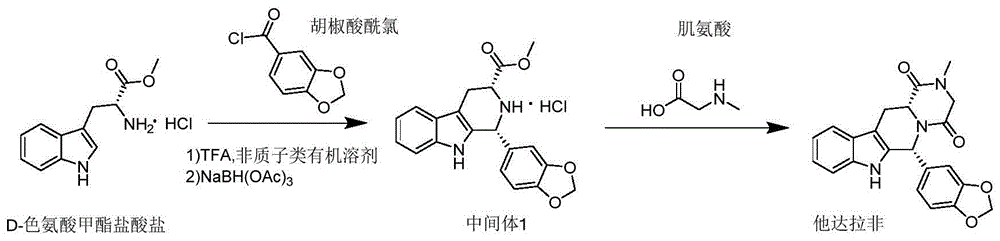
目前,现有技术主要以d-色氨酸甲酯、d-色氨酸甲酯盐酸盐、d-色氨酸、nα-boc-d-色氨酸为原料与胡椒醛或其类似物反应,经过一系列的合成步骤制备他达拉非。如us2009312548公开了一种他达拉非的制备方法,采用d-色氨酸经二氯亚砜甲酯化与胡椒醛缩合,再接氯乙酰氯之后跟甲胺环合,制备得到他达拉非。但该方法所采用的胡椒醛为管制类药物不易获得,且二氯亚砜、氯乙酰氯、甲胺等试剂存在毒性较大、易燃易爆、环境污染严重等问题。
为了解决上述问题,伍普华等人公开了一种他达拉非的合成路线:
在上述合成路线中,先将d-色氨酸甲酯盐酸盐与胡椒酸酰氯缩合制得化合物9,经劳森试剂作用制得化合物10,再经过碘甲烷回流、硼氢化钠还原环化得到化合物7(中间体1),收率为62.5%,然后再与氯乙酰氯进行三乙胺催化缩合、甲胺乙醇关环反应制备得到终产物他达拉非,收率为66.4%(伍普华,谢义鹏.他达拉非合成路线图解[j].中国药物化学杂志,2015,25(05):407-410.)。虽然该方法避免了使用管制类原料胡椒醛、有毒有害试剂二氯亚砜等,但是该方法步骤繁多,容易产生多种副产物,且各步骤收率较低,最后总收率仅为25.9%,再加上反应过程中使用的劳森试剂气味难闻,不适合实际生产。
因此,迫切需要提供一种避免使用管制类、有毒有害类试剂,步骤简单,收率高,纯度好的他达拉非制备方法。
技术实现要素:
本发明要解决的技术问题是克服现有技术反应中常使用管制类、有毒有害类试剂,步骤繁琐,收率低等缺陷和不足,提供一种避免使用管制类、有毒有害类试剂,步骤简单,收率高,纯度好的他达拉非制备方法。
本发明上述目的通过以下技术方案实现:
一种他达拉非的制备方法,反应路线如下:
具体包括以下步骤:
s1.将d-色氨酸甲酯盐酸盐和胡椒酸酰氯溶于非质子类有机溶剂中,加入三氟乙酸,在氮气保护下,30~45℃搅拌反应8~12h,再加入三乙酰氧基硼氢化钠,继续反应2~4h,得含有中间体1的反应液;
s2.向步骤s1.所得含有中间体1的反应液中加入肌氨酸和三乙胺,20~35℃搅拌15~25min,缓慢滴加n,n'-二环己基碳酰亚胺,加入4-二甲氨基吡啶,混合反应7~8h,冷却至2~5℃,加入碱性试剂,回流反应8~10h,后处理,分离,得他达拉非;
其中,步骤s1.中,所述d-色氨酸甲酯盐酸盐和三氟乙酸的物质的量比为1:(0.02~0.5);
步骤s2.中,所述碱性试剂选自三乙胺、n,n-二异丙基乙胺、甲醇镁、氢氧化钠、氢氧化钾中的一种。
其中,步骤s1.的反应机理可能为:
首先,d-色氨酸甲酯盐酸盐与胡椒酸酰氯在微量酸性催化剂三氟乙酸的作用下先进行傅克-酰基化反应,再进行席夫碱的反应,形成的亚胺化合物不稳定,加入三乙酰氧基硼氢化钠进一步还原胺化形成中间体1。整个反应过程一气呵成,无需分离中间产物,通过添加三乙酰氧基硼氢化钠控制反应的进程,可以看成一步反应制备得到中间体1。
其中,傅克-酰基化反应中常采用路易斯酸(如三氯化铝)进行催化,但该类催化剂在反应过程中容易生成氢氧化物的胶体,不仅会覆盖在反应物的表面,影响反应的进程,还容易堵塞滤纸,不易过滤;而且,为了保证傅克-酰基化反应的正常进行,一般还需要添加大量的金属卤化物,成本较高。申请人在实践中惊喜的发现,采用催化量的质子酸三氟乙酸,不需要再加入别的试剂,即可同时催化傅克-酰基化反应和席夫碱反应,在一定的时间内,可以反应形成亚胺化合物,经过还原后所得中间体1具有较高的收率和纯度,符合绿色化学的思想。
步骤s2.中,本发明采用肌氨酸与中间体1先进行酰胺化反应,然后在碱性试剂条件下,回流反应进行胺解反应,通过对碱性试剂、回流温度等条件的控制,可以得到高收率的他达拉非,解决了采用肌氨酸酯盐酸盐或肌氨酸酯反应的反应速率低、反应时间长、收率低等问题。
优选的,步骤s1.中,所述d-色氨酸甲酯盐酸盐和三氟乙酸的物质的量比为1:(0.05~0.2)。
更优选的,步骤s1.中,所述d-色氨酸甲酯盐酸盐和三氟乙酸的物质的量比为1:0.05。申请人在实践中发现,当d-色氨酸甲酯盐酸盐和三氟乙酸的物质的量比为1:0.05时,中间体1的收率较高。
优选的,步骤s1.中,所述反应的温度为30~40℃。
更优选的,步骤s1.中,所述反应的温度为30℃。申请人在实践中发现,当所述反应的温度为30℃时,中间体1的收率较高。
进一步地,步骤s2.中,所述回流反应的温度为50~80℃。
优选的,步骤s2.中,所述回流反应的温度为60~80℃。
更优选的,步骤s2.中,所述回流反应的温度为80℃。申请人在实践中发现,当所述回流反应的温度为80℃时,他达拉非的收率较高。
优选的,步骤s2.中,所述碱性试剂选自三乙胺、n,n-二异丙基乙胺、甲醇镁中的一种。
更优选的,步骤s2.中,所述碱性试剂为甲醇镁。申请人在实践中发现,当所述碱性试剂为甲醇镁时,他达拉非的收率较高。
更进一步地,步骤s1.中,所述d-色氨酸甲酯盐酸盐和三乙酰氧基硼氢化钠的物质的量比为1:(1~5)。
优选的,步骤s1.中,所述d-色氨酸甲酯盐酸盐和三乙酰氧基硼氢化钠的物质的量比为1:(3~5)。
更优选的,步骤s1.中,所述d-色氨酸甲酯盐酸盐和三乙酰氧基硼氢化钠的物质的量比为1:3。
进一步地,步骤s2.中,所述中间体1、碱性试剂的物质的量比为1:(1~3)。
优选的,步骤s2.中,所述中间体1、碱性试剂的物质的量比为1:(1~2)。
更优选的,步骤s2.中,所述中间体1、碱性试剂的物质的量比为1:1.5。
更进一步地,步骤s2.中,所述混合反应的温度为20~35℃。
优选的,步骤s2.中,所述混合反应的温度为30~35℃。
更优选的,步骤s2.中,所述混合反应的温度为30℃。
进一步地,步骤s2.中,所述中间体1、n,n'-二环己基碳酰亚胺、4-二甲氨基吡啶的物质的量比为1:(1~3):(0.005~0.02)。
优选的,步骤s2.中,所述中间体1、n,n'-二环己基碳酰亚胺、4-二甲氨基吡啶的物质的量比为1:(2~3):(0.01~0.02)。
更优选的,步骤s2.中,所述中间体1、n,n'-二环己基碳酰亚胺、4-二甲氨基吡啶的物质的量比为1:2:0.01。
更进一步地,步骤s1.、s2.中,所述非质子类有机溶剂选自四氢呋喃、甲苯、二氯甲烷、n,n二甲基甲酰胺、乙腈、二甲基亚砜中的一种。
优选的,步骤s1.、s2.中,所述非质子类有机溶剂为四氢呋喃。
本发明具有以下有益效果:
本发明一种他达拉非的制备方法,以较为容易获得的胡椒酸酰氯等为原料,制备得到的中间体1与肌氨酸进行酰胺化反应,在特定的反应条件下,可以高收率的制备得到高纯度的他达拉非,并且,整个合成过程仅分为两步,可以通过一锅煮的方式进行制备,与现有技术相比,显著减少了复杂的反应步骤,操作简单,适合大规模产业化生产他达拉非。
附图说明
图1为本发明他达拉非制备方法的合成路线图。
图2为本发明实施例1制备的他达拉非的氢谱图。
图3为本发明实施例1制备的他达拉非的质谱图。
图4为本发明实施例1制备的他达拉非的高效液相色谱图。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
本发明他达拉非制备方法的合成路线如下所示:
实施例1他达拉非的制备
采用如下方法制备分离得到所述他达拉非:
s1.将9.34gd-色氨酸甲酯盐酸盐和8.12g胡椒酸酰氯溶于300ml重蒸的四氢呋喃中,加入0.14ml三氟乙酸,在氮气保护下,30℃搅拌反应10h,再加入23.30g三乙酰氧基硼氢化钠,继续反应3h,tlc板监测反应结束后,得300ml含有12.33g中间体1的四氢呋喃溶液;
s2.向步骤s1.所得含有12.33g中间体1的四氢呋喃溶液中加入3.41g肌氨酸和3.23g三乙胺,30℃搅拌20min,缓慢滴加30ml含13.16gn,n'-二环己基碳酰亚胺的四氢呋喃溶液,30min滴加完毕后,加入0.04g4-二甲氨基吡啶,30℃混合反应7h,反应结束后,将反应液冷却至4℃,加入7wt%含有4.13g甲醇镁的甲醇溶液,升温至80℃回流反应9h,tlc板监测反应结束后,冷却至室温,加入15g硅藻土,混合、过滤,固体残渣回收、洗涤,滤液减压浓缩至30ml左右,加入100ml去离子水,用200ml乙酸乙酯分两次萃取,合并有机相,重结晶4次,固体物质在25℃真空干燥,得11.24g他达拉非,收率90.54%,纯度100.00%,esi(m/z)[m+h]+:390.1463,[m+na]+:412.1279。
实施例2三氟乙酸添加量、温度、溶剂对中间体1收率的影响
参考实施例1他达拉非的制备,不同之处在于,在步骤s1.中,采用不同的三氟乙酸添加量、温度、溶剂制备中间体1,计算所得中间体1的收率。具体三氟乙酸添加量、温度、溶剂和实验结果参见表1。
表1三氟乙酸添加量、温度、溶剂对中间体1收率的影响
由表1可见:
(1)本发明步骤s1.中,在反应温度和溶剂相同条件下,当d-色氨酸甲酯盐酸盐和三氟乙酸的物质的量比在1:(0.05~0.2)之间时,中间体1的收率在75%以上,收率较高,当超出此范围时,中间体1的收率显著降低至50%以下;
(2)本发明步骤s1.中,在物质的量比和试剂相同条件下,制备得到的中间体1的收率随反应温度的升高而降低,其中,当反应温度为30~40℃时,收率可达70%以上;
(3)本发明步骤s1.中,在物质的量比和温度相同条件下,选用不同的溶剂也会对中间体1的收率产生一定的影响,其中,当溶剂为四氢呋喃时,收率最高。实施例3碱性试剂、回流反应温度、非质子类有机溶剂对他达拉非收率的影响
参考实施例1他达拉非的制备,不同之处在于,在步骤s2.中,采用不同的碱性试剂、回流反应温度、非质子类有机溶剂制备他达拉非,计算所得他达拉非的收率。具体碱性试剂、回流反应温度、非质子类有机溶剂和实验结果参见表2。
表2碱性试剂、回流反应温度、非质子类有机溶剂对他达拉非收率的影响
由表2可见:
(1)本发明步骤s2.中,在相同温度和溶剂条件下,除了碳酸钾只能制备得到微量他达拉非以外,其余采用的碱性试剂均能制备得到一定量的他达拉非;而其中,甲醇镁制备得到的他达拉非收率最高;
(2)本发明步骤s2.中,在相同碱性试剂和溶剂条件下,回流反应温度50~80℃都能制备得到一定量的他达拉非,但回流反应温度对他达拉非的收率大小有一定的影响,其中,当回流反应温度为80℃时,他达拉非的收率最高;
(3)本发明步骤s2.中,在相同碱性试剂和温度条件下,选用不同的溶剂也会对他达拉非的收率产生一定的影响,其中,当溶剂为四氢呋喃时,收率最高。实验例1他达拉非的氢谱、质谱和高效液相色谱检测
对实施例1制备的他达拉非进行氢谱、质谱和高效液相色谱检测,得图2~4。
其中,高效液相色谱的检测方法为:
取实施例1制备的他达拉非适量,精密称定,参考《中国药典》(2015年版)第四部通则0512高效液相色谱法,用十八烷基硅烷键合硅胶为填充剂,设置检测波长为285nm,流速为1.0ml/min,进样量10μl,以0.1%的三氟乙酸为流动相a,以乙腈为流动相b,按表3条件进行梯度洗脱,记录色谱图。
表3高效液相色谱梯度洗脱条件
由图4可知,他达拉非的出峰时间在24.141min。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!