恩格列净的合成方法与流程
本发明属于医药和精细化工领域。具体地说,本发明涉及恩格列净的新合成方法以及用于合成恩格列净的新中间体。
背景技术:
恩格列净(empagliflozin)是勃林格殷格翰司开发的选择性口服sglt-2抑制药,是一种新型的口服降糖药。2014年5月率先在欧洲上市,2014年8月和12月分别在美国和日本上市,并于2017年9月在中国上市。恩格列净能够通过选择性抑制肾小球近端小管对滤过葡萄糖重吸收,使过量的葡萄糖从尿液中排出,直接降低血糖,用于治疗ii型糖尿病,临床研究结果表明疗效显著,并具有很好的安全性和耐受性。
恩格列净化学名为:(1s)-1,5-脱水-1-c-(4-氯-3-((4-((((3s)-四氢-3-呋喃基)氧基)苯基)甲基)苯基)-d-葡萄糖醇。恩格列净结构如下:
关于恩格列净的合成,现有技术中已有多篇文献报道。例如,wo2006120208公开了一种恩格列净的制备方法,具体的合成过程如下所示:在-78℃下,将(s)-4-溴-1-氯-2-(4-四氢呋喃-3-基氧基-苄基)-苯在无水乙醚中和叔丁基锂进行卤素-金属交换;随后加入2,3,4,6-四-o-(三甲基硅烷基)-d-葡萄吡喃酮进行加成反应;反应完毕后,添加甲醇溶液和甲磺酸溶液继续反应生成1-氯-4-(1-甲氧基-d-吡喃葡萄糖-1-基)-2-(4-(s)-四氢呋喃-3-基氧基-苄基)-苯;最后在三乙基甲硅烷和三氟化硼乙醚的作用下经还原得到恩格列净。
此方法要求在低温(-78℃)下进行,操作繁琐,生成的锂盐与保护的葡糖糖内酯反应不完全会产生以下所示的式10杂质和开环杂质式11,而且2,3,4,6-四-o-(三甲基硅烷基)-d-葡萄吡喃酮不稳定容易水解。因此,该路线的总收率非常低,仅为12%左右,工艺操作成本较高。
j.labelcompd.radiopharm.2014期,57卷,687页报道了恩格列净的一种制备方法。其中,氟苯与(s)-3-羟基-四氢呋喃反应,再与酰氯化试剂发生傅克反应,然后用et3sih/bf3·et2o体系进行还原,再用正丁基锂拔卤后形成锂盐与葡萄糖酯发生亲核加成反应,然后在用甲醇进行甲醚化,最后用et3sih/bf3·et2o体系还原得到恩格列净。
此方法需要用到易自燃的试剂,正丁基锂,并且反应温度同样苛刻(-78℃)。此外,该方法同样有副反应发生(生成的锂盐与保护的葡糖糖内酯反应不完全有式10杂质和开环杂质式11),而且2,3,4,6-四-o-(三甲基硅烷基)-d-葡萄吡喃酮不稳定容易水解。因此,该方法的总收率依然很低,仅有13.5%左右,从而造成该路线的成本仍然偏高。
因此,本领域急需一种恩格列净的合成方法,该方法应具备高收率、低成本以及操作方便、适合大工业化生产等优点。
技术实现要素:
本发明的目的在于提供一种恩格列净的合成方法,该方法能够以高收率、低成本地制备恩格列净,并且同时具备操作方便、适合大工业化生产等优点。
在第一方面,本发明提供如下所示的恩格列净合成方法,包含以下步骤:
1)在有碱和催化剂存在下,(s)-4-卤代-1-氯-2-(4-四氢呋喃-3-基氧基-苄基)-苯(式5化合物)与联硼酸频哪醇酯(式6化合物)发生偶联反应得到(s)-2-(4-氯-3-(4-((四氢呋喃-3-基)甲基)苄基)苯基)-4,4,5,5-四甲基-1,3,2-二噁硼烷(式7化合物);
2)在有碱和催化剂存在下,将(s)-2-(4-氯-3-(4-((四氢呋喃-3-基)甲基)苄基)苯基)-4,4,5,5-四甲基-1,3,2-二噁硼烷(式7化合物)与乙酰溴-α-d-葡萄糖(式8化合物)发生偶联反应得到1-氯-4-(2,3,4,6-四-o-乙酰基-d-吡喃葡萄糖-1-基)-2-(4-(s)-四氢呋喃-3-基氧基-苄基)-苯(式9化合物);和
3)将1-氯-4-(2,3,4,6-四-o-乙酰基-d-吡喃葡萄糖-1-基)-2-(4-(s)-四氢呋喃-3-基氧基-苄基)-苯(式9化合物)脱保护得到恩格列净。
在具体的实施方式中,步骤1)在单一溶剂或组合溶剂中进行;包括但不限于甲苯/甲苯水两相溶液、四氢呋喃/四氢呋喃水溶液、乙腈/乙腈水溶液、二氧六环/二氧六环水溶液、n,n-二甲基甲酰胺(dmf)/dmf乙醇混合溶液/dmf甲醇溶液、n,n-二甲基乙酰胺(dmac)、dmso/dmso水溶液、甲基吡咯烷酮(nmp),优选二氧六环水溶液组合溶剂。
在具体的实施方式中,步骤1)中的碱为有机碱或无机碱;其中有机碱包括但不限于三乙胺、n,n-二异丙基乙胺、甲醇钠(钾)、乙醇钠(钾)、叔丁醇钠(钾)、醋酸钠(钾);无机碱包括但不限于碳酸钠、碳酸钾、碳酸铯、磷酸钾、氢氧化钠、氢氧化钾,优选醋酸钾。
在具体的实施方式中,步骤1)中碱的用量相对于式5化合物的摩尔比为:2eq-5eq,优选3eq-3.5eq。
在具体的实施方式中,步骤1)中的催化剂为钯或镍化合物;所述镍化合物包括但不限于氯化镍、溴化镍、1,1'-双二苯基膦二茂铁二氯化镍(dppfnicl2)、双(三苯基膦)氯化镍nicl2(pph3)2,优选dppfnicl2;钯化合物包括但不限于氯化钯、醋酸钯、四三苯基膦钯、1,1'-双二苯基膦二茂铁二氯化钯(dppfpdcl2)、双(三苯基膦)氯化钯pdcl2(pph3)2、pd(dba)2,优选dppfpdcl2。
在具体的实施方式中,步骤1)中的催化剂用量为:0.5mol%-10mol%,优选2mol%-5mol%。
在具体的实施方式中,步骤1)中的温度为80-140℃,优选95-100℃。
在具体的实施方式中,步骤2)中的碱为有机碱或无机碱;所述有机碱包括但不限于三乙胺、n,n-二异丙基乙胺、甲醇钠(钾)、乙醇钠(钾)、叔丁醇钠(钾)、醋酸钠(钾);所述无机碱包括但不限于碳酸钠、碳酸钾、碳酸铯、磷酸钾、氢氧化钠、氢氧化钾,优选磷酸钾。
在具体的实施方式中,步骤2)中碱的用量为:2eq-6eq,优选3eq-4eq。
在具体的实施方式中,步骤2)中的催化剂为钯或镍化合物;镍化合物为氯化镍、溴化镍、四三苯基膦镍、1,1'-双二苯基膦二茂铁二氯化镍(dppfnicl2)、双(三苯基膦)氯化镍[nicl2(pph3)2)],优选四三苯基膦镍;钯化合物为氯化钯、醋酸钯、四三苯基膦钯、1,1'-双二苯基膦二茂铁二氯化钯(dppfpdcl2)、双(三苯基膦)氯化钯pdcl2(pph3)2、pd(dba)2,优选四三苯基膦钯。
在具体的实施方式中,步骤2)中的催化剂用量为:0.01mol%-5mol%,优选1mol%-2mol%。
在具体的实施方式中,步骤2)在单一溶剂或组合溶剂中进行;包括但不限于甲苯/甲苯水两相溶液、四氢呋喃/四氢呋喃水溶液、乙腈/乙腈水溶液、二氧六环/二氧六环水溶液、n,n-二甲基甲酰胺(dmf)/dmf乙醇混合溶液/dmf甲醇溶液、n,n-二甲基乙酰胺(dmac)、dmso/dmso水溶液、甲基吡咯烷酮(nmp),优选甲苯水两相溶液。
在具体的实施方式中,步骤2)中的温度为70-140℃,优选100-110℃。
在优选的实施方式中,所述恩格列净合成方法的收率为20%以上,优选25%以上。更优选30%以上。
在第二方面,本发明提供下式7所示化合物,
在第三方面,本发明提供式7所示化合物的制备方法,如以下反应流程所示:
在有碱和催化剂存在下,(s)-4-卤代-1-氯-2-(4-四氢呋喃-3-基氧基-苄基)-苯(式5化合物)与联硼酸频哪醇酯(式6化合物)发生偶联反应得到(s)-2-(4-氯-3-(4-((四氢呋喃-3-基)甲基)苄基)苯基)-4,4,5,5-四甲基-1,3,2-二噁硼烷(式7化合物)。
在优选的实施方式中,所示方法在单一溶剂或组合溶剂中进行;包括但不限于甲苯/甲苯水两相溶液、四氢呋喃/四氢呋喃水溶液、乙腈/乙腈水溶液、二氧六环/二氧六环水溶液、n,n-二甲基甲酰胺(dmf)/dmf乙醇混合溶液/dmf甲醇溶液、n,n-二甲基乙酰胺(dmac)、dmso/dmso水溶液、甲基吡咯烷酮(nmp),优选二氧六环水溶液组合溶剂。
在优选的实施方式中,所示方法中的碱为有机碱或无机碱;有机碱包括但不限于三乙胺、n,n-二异丙基乙胺、甲醇钠(钾)、乙醇钠(钾)、叔丁醇钠(钾)、醋酸钠(钾);无机碱包括但不限于碳酸钠、碳酸钾、碳酸铯、磷酸钾、氢氧化钠、氢氧化钾,优选醋酸钾。
在优选的实施方式中,所示方法中碱的用量为:2eq-5eq,优选3eq-3.5eq。
在优选的实施方式中,所示方法中的催化剂为钯或镍化合物;所述镍化合物包括但不限于氯化镍、溴化镍、1,1'-双二苯基膦二茂铁二氯化镍(dppfnicl2)、双(三苯基膦)氯化镍nicl2(pph3)2,优选dppfnicl2;钯化合物包括但不限于氯化钯、醋酸钯、四三苯基膦钯、1,1'-双二苯基膦二茂铁二氯化钯(dppfpdcl2)、双(三苯基膦)氯化钯pdcl2(pph3)2、pd(dba)2,优选dppfpdcl2。
在优选的实施方式中,所示方法中的催化剂用量为:0.5mol%-10mol%,优选2mol%-5mol%。
在优选的实施方式中,所示方法中的温度为80-140℃,优选95-100℃。
在第四方面,本发明提供式7所示化合物在制备恩格列净中的用途。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
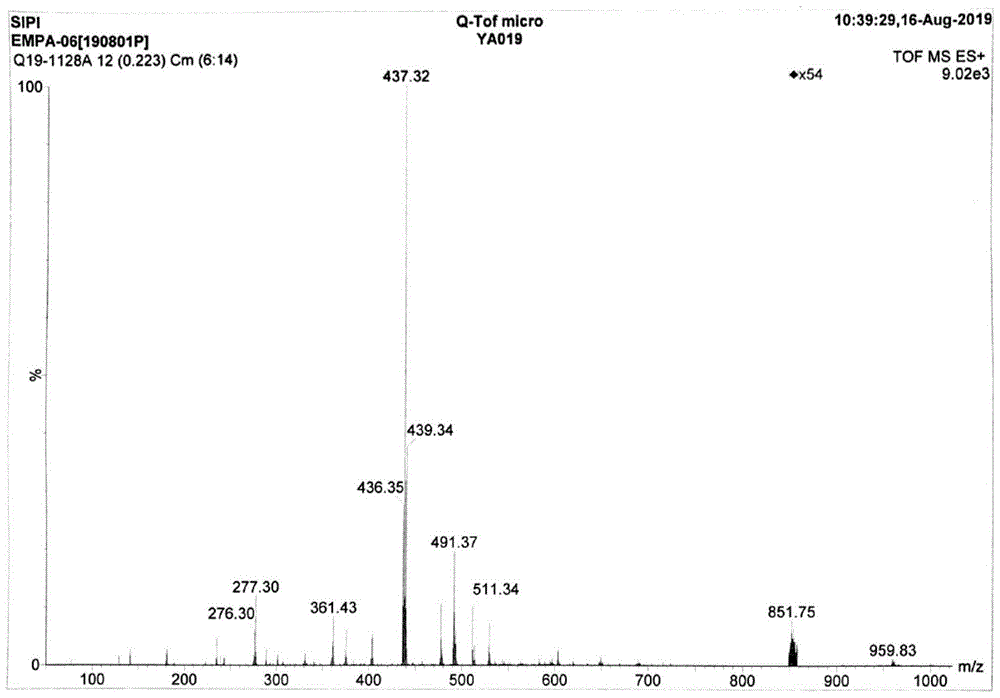
图1显示了式7化合物的质谱图;
图2显示了式7化合物的hnmr图;
图3显示了恩格列净的质谱图;
图4显示了恩格列净的hnmr图。
具体实施方式
发明人经过广泛而深入的研究,出乎意料地发现了一种全新的恩格列净合成工艺。该工艺利用硼酸酯进行拔卤作用,再结合特定的反应条件从而能够高收率、操作简便地制备恩格列净。在此基础上完成了本发明。
为克服现有的技术中的不足,本发明提供了一种高效的合成恩格列净的方法,其合成路线如下所示(涉及以下3个反应步骤):
步骤1、在有碱和催化剂存在下,(s)-4-卤代-1-氯-2-(4-四氢呋喃-3-基氧基-苄基)-苯(式5化合物)与联硼酸频哪醇酯(式6化合物)发生偶联反应得到(s)-2-(4-氯-3-(4-((四氢呋喃-3-基)甲基)苄基)苯基)-4,4,5,5-四甲基-1,3,2-二噁硼烷(式7化合物);
步骤2、在有碱和催化剂存在下,将(s)-2-(4-氯-3-(4-((四氢呋喃-3-基)甲基)苄基)苯基)-4,4,5,5-四甲基-1,3,2-二噁硼烷(式7化合物)与乙酰溴-α-d-葡萄糖(式8化合物)发生偶联反应得到1-氯-4-(2,3,4,6-四-o-乙酰基-d-吡喃葡萄糖-1-基)-2-(4-(s)-四氢呋喃-3-基氧基-苄基)-苯(式9化合物);和
步骤3、将1-氯-4-(2,3,4,6-四-o-乙酰基-d-吡喃葡萄糖-1-基)-2-(4-(s)-四氢呋喃-3-基氧基-苄基)-苯(式9化合物)脱保护得到恩格列净。
此方法减少了副反应的发生,相比之下,现有技术方法,例如wo2006120208和j.labelcompd.radiopharm所述的方法均有副反应发生,从而生成杂质10和杂质11。此外,本发明方法的反应条件温和,总收率高(可达20%以上,优选25%以上。更优选30%以上),从而有利于工业化生产。
根据本发明的教导,本领域技术人员可以凭借现有技术知识知晓如何获得上述工艺中的一些中间体。例如,在本发明的方法中,(s)-4-卤代-1-氯-2-(4-四氢呋喃-3-基氧基-苄基)-苯(式5)可通过已知方法,例如wang,xiao-jun;zhang,li;organicletters,2014,vol.16,#16p.4090-4093以及wo2011039107,us2011237789等描述的方法制得。例如,价廉易得的2-氯-5卤代苯甲酸(式1)首先酰化得到式2,再与氟苯发生傅克反生成式3,然后与(s)-3-羟基-四氢呋喃反应生成式4,最后用硼氢化钠/三氯化铝作用下还原得到(s)-4-卤代-1-氯-2-(4-四氢呋喃-3-基氧基-苄基)-苯(式5)。
然而,在进一步的研究中,本发明人发现以上步骤1和2中采用的具体反应物和工艺参数对中间体式7化合物和式9化合物以及最终恩格列净的收率亦有相当影响。
例如,在步骤1中,宜利用单一溶剂或组合溶剂进行反应;例如甲苯/甲苯水两相溶液、四氢呋喃/四氢呋喃水溶液、乙腈/乙腈水溶液、二氧六环/二氧六环水溶液、n,n-二甲基甲酰胺(dmf)/dmf乙醇混合溶液/dmf甲醇溶液、n,n-二甲基乙酰胺(dmac)、dmso/dmso水溶液、甲基吡咯烷酮(nmp),优选二氧六环水溶液组合溶剂。利用的碱宜是有机碱或无机碱,例如三乙胺、n,n-二异丙基乙胺、甲醇钠(钾)、乙醇钠(钾)、叔丁醇钠(钾)、醋酸钠(钾)等有机碱;或者碳酸钠、碳酸钾、碳酸铯、磷酸钾、氢氧化钠、氢氧化钾等无机碱;优选醋酸钾。碱的用量相对于式5化合物的摩尔比宜为:2eq-5eq,优选3eq-3.5eq。催化剂宜用钯或镍化合物,例如氯化镍、溴化镍、1,1'-双二苯基膦二茂铁二氯化镍(dppfnicl2)、双(三苯基膦)氯化镍nicl2(pph3)2,优选dppfnicl2;或氯化钯、醋酸钯、四三苯基膦钯、1,1'-双二苯基膦二茂铁二氯化钯(dppfpdcl2)、双(三苯基膦)氯化钯pdcl2(pph3)2、pd(dba)2,优选dppfpdcl2;而催化剂的用量可以为:0.5mol%-10mol%,优选2mol%-5mol%;步骤1的反应宜在80-140℃,优选95-100℃的温度下进行。
在另一具体的实施方式中,在步骤2中,碱宜用三乙胺、n,n-二异丙基乙胺、甲醇钠(钾)、乙醇钠(钾)、叔丁醇钠(钾)、醋酸钠(钾)等有机碱或碳酸钠、碳酸钾、碳酸铯、磷酸钾、氢氧化钠、氢氧化钾等无机碱,优选磷酸钾。碱的用量宜为2eq-6eq,优选3eq-4eq。催化剂宜用钯或镍化合物;镍化合物为氯化镍、溴化镍、四三苯基膦镍、1,1'-双二苯基膦二茂铁二氯化镍(dppfnicl2)、双(三苯基膦)氯化镍[nicl2(pph3)2)],优选四三苯基膦镍;钯化合物为氯化钯、醋酸钯、四三苯基膦钯、1,1'-双二苯基膦二茂铁二氯化钯(dppfpdcl2)、双(三苯基膦)氯化钯pdcl2(pph3)2、pd(dba)2,优选四三苯基膦钯。催化剂的用量宜为0.01mol%-5mol%,优选1mol%-2mol%。步骤2的反应宜在单一溶剂或组合溶剂中进行;例如甲苯/甲苯水两相溶液、四氢呋喃/四氢呋喃水溶液、乙腈/乙腈水溶液、二氧六环/二氧六环水溶液、n,n-二甲基甲酰胺(dmf)/dmf乙醇混合溶液/dmf甲醇溶液、n,n-二甲基乙酰胺(dmac)、dmso/dmso水溶液、甲基吡咯烷酮(nmp),优选甲苯水两相溶液。步骤2的反应宜在70-140℃,优选100-110℃的温度下进行。
本发明的主要优点包括:
1.本发明方法合成恩格列净的反应条件温和;
2.本发明方法合成恩格列净的总收率高;
3.本发明方法合成恩格列净的副反应少,从而不宜产生杂质;
4.本发明方法合成恩格列净的操作方便,从而有利于工业化生产和成本控制。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
实施例1.合成((2-氯-5-溴苯基)(4-氟苯基)甲酮)(式3)
合成式2:反应瓶中加入235.5g的式1;1l的二氯甲烷;10毫升的dmf。降温至内温10℃以下。滴加入140g的草酰氯。滴加完毕后,控制温度在20℃以下反应120min,tlc检测反应。浓缩除去反应溶剂二氯甲烷和过量的草酰氯得到252.8g的白色固体式2。
合成式3:反应瓶中加入1l的二氯甲烷,再加入190g的氟苯。冰水降温至0℃以下。控温加入185g的无水三氯化铝。加完后滴加入式2(252.8g溶于1l的二氯甲烷)。加完后室温反应5小时,tlc检测反应完全。反应完倒入冰水中。用饱和氯化钠洗两次至中性,无水硫酸镁干燥,过滤浓缩得到的固体用乙酸乙酯精制得到白色固体272.6g。收率86.9%。
实施例2.合成[4-[[(3s)-四氢-3-呋喃]氧基]苯基]-(5-溴-2-氯苯基)甲酮(式4)
反应瓶中加入250g的式3化合物,然后加入750ml的四氢呋喃溶解,再加入78g的(s)-3-羟基四氢呋喃。冰水降温0℃以下。控温加入叔丁醇钾溶液(122g的叔丁醇钾溶于1l的四氢呋喃中)。加完后在10℃以下反应2小时,tlc检测反应完全。反应液倒入冰水中搅拌加入异丙醚萃取。用饱和氯化钠洗两次至中性,无水硫酸镁干燥,过滤浓缩得到残余物用丙酮精制得到259.7g的白色固体式4化合物,收率85.3%。
实施例3.合成(s)-3-(4-(5-溴-2-氯苄基)苯氧基)四氢呋喃(式5)
反应瓶中加入247g的式4化合物,2l的四氢呋喃。降温至内温20℃以下。控制温度加入159g的硼氢化钾。加完后控温20℃以下分批加入193g的无水三氯化铝。加完后回流反应6小时,tlc检测反应全。反应液倒入冰水中。滴加稀盐酸。乙酸乙酯萃取,干燥浓缩得到残余物用乙醇精制得到193.5g的白色式5化合物,收率81.8%。
实施例4.合成(s)-2-[4-氯-3-(4-((四氢-3-呋喃基)氧基)苯基)苯基)-4,4,5,5-四甲基-1,3,2-二噁硼烷(式7)化合物
反应瓶中加入36.6g的式5化合物,350毫升的二氧六环,38g的联硼酸频那醇酯(式6),30g的醋酸钾,1.47g的dppfpdcl,氮气填充下油浴升温至100℃反应8小时,反应完后降至室温,过滤,滤液水洗后浓缩得到黄色固体加入乙醇精制得到白色固体(式7),26.1g。收率71.3%。hplc:98.6%。
esi-msm/z:437.32[m+na]+:1h-nmr(400mhz,cdcl3)δ:1.25~127(t,12h),2.03-2.09(m,2h),3.13(s,1h),3.78~3.99(m,4h),3.97(s,2h),6.67~6.69(d,2h),7.27~7.29(d,1h),7.51~7.53(d,1h),7.61(s,1h)。
实施例5.合成1-氯-4-(2,3,4,6-四-o-乙酰基-d-吡喃葡萄糖-1-基)-2-(4-(s)-四氢呋喃-3-基氧基-苄基)-苯(式9)
式8化合物合成:反应瓶中加入葡萄糖40g,然后加入200ml醋酸酐。加完后升温至溶解。控温加入醋酸钠10g。加完后140℃以下反应2小时,tlc检测反应完全。反应液倒入冰水中搅拌过滤水洗得到的粗品用乙醇精制得到白色固体β-d-葡萄糖五乙酸酯80g。
将上述的得到的β-d-葡萄糖五乙酸酯80g加入醋酸酐400毫升,三溴化磷120ml。降温至内温0℃以下。控制温度滴加入175g的水滴加完毕控温0℃反应60min,tlc检测反应。反应液倒入冰水中。二氯甲烷萃取,有机层水洗,碳酸氢钠溶液洗,再饱和食盐水洗,干燥浓缩得到残余物用异丙醚精制得到的白色固体式8化合物,72.5g。
式9化合物合成:反应瓶中加入(式7)55g,甲苯500毫升,式8化合物72.5g,磷酸钾85g,四三苯基膦钯5g,油浴升温至105℃反应12小时,反应完后降至室温,过滤,滤液用250毫升×2水洗两次,有机层浓缩得到残余液用乙酸乙酯精制得到式9化合物63.5g,收率77.4%,hplc:99.4%。
esi-msm/z:641.11[m+na]+。
1h-nmr(400mhz,cdcl3)δ:2.49~2.51(m,9h),3.32(s,18h)。
实施例6.合成恩格列净
反应瓶中加入式9化合物31g,甲醇50毫升,搅拌溶解后控制20℃以下滴加20%的氢氧化钠溶液50g,滴加完毕后反应液在20℃以下反应60分钟,hplc检测反应完后用稀盐酸调ph=7,浓缩甲醇,残余液用二氯甲烷萃取两次,合并有机层浓缩,残余液加入乙酸乙酯和水精制得到20.7g,本步骤收率91.7%,hplc=99.1%。
esi-msm/z:473.21.[m+na]+。
1h-nmr(400mhz,cdcl3)δ:lhnmr(dms0-d6)б:7.37(d,j=8.2hz,1h),7.33(d,j=1.9hz,1h),7.23(dd,j=8.3,2.1hz,1h),7.10(d,j=8.6hz,2h),6.82(d,j=8.7hz,2h),4.94(m,3h),4.82(d,j=5.8hz,1h),4.43(t,j=5.8hz,1h),4.04(m,3h),3.84(m,2h),3.75(m,3h),3.43(m,1h),3.30(m,3h),2.21(m,1h),1.95(m,1h)。
实施例7:合成式7化合物
不同催化剂c1对比研究:参照实施例4中的实验操作,只改变投料参数中的催化剂种类,其余的参数不变经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表1。
表1
实施例8:合成式7化合物
催化剂用量对比研究:合成实验组参照实施例4中的实验操作,只改变投料参数中的催化剂的用量(相对式5的原料摩尔比),其余的参数不变。经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表2。
表2
实施例9:合成式7化合物
不同溶剂s1对比研究:合成实验组参照实施例4中的实验操作,只改变投料参数中的s1溶剂类型,其余的参数不变。经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表3。
表3
实施例10:合成式7化合物
不同碱b1对比研究:合成实验组参照实施例4中的实验操作,只改变投料参数中的b1碱种类型,其余的参数不变。经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表4。
表4
实施例11:合成式7化合物
不同碱用量对比研究:合成实验组参照实施例4中的实验操作,只改变投料参数中的b1碱种(醋酸钾)用量(相对式5的原料摩尔比),其余的参数不变。经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表5。
表5
实施例11:合成式7化合物
不同溶剂用量对比研究:合成实验组参照实施例4中的实验操作,只改变投料参数中的s1溶剂(二氧六环)用量(相对式5的质量比),其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表6。
表6
实施例12:合成式7化合物
不同温度t1对比研究:合成实验组参照实施例4中的实验操作(反应溶剂为dmf),只改变反应过程的t1温度参数中的t1。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式7化合物的纯度和收率见表7。
表7
实施例13:合成式9化合物
不同催化剂c2对比:合成实验组参照实施例5中的式9化合物的实验操作,只改变投料过程的c2催化剂。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表8。
表8
实施例14:合成式9化合物
催化剂用量对比研究:合成实验组参照实施例5中的式9化合物的实验操作,只改变投料过程的c2催化剂(四三苯基膦钯)的用量(相对式7的原料摩尔比)。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表9。
表9
实施例15:合成式9化合物
不同溶剂s2对比研究:合成实验组参照实施例5中的式9化合物的实验操作,只改变投料过程的s2溶剂的类型。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表10。
表10
实施例16:合成式9化合物
不同碱b2对比研究:合成实验组参照实施例5中的式9化合物的实验操作,只改变投料过程的b2碱的类型。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表11。
表11
实施例17:合成式9化合物
不同碱用量对比研究:合成实验组参照实施例5中的式9化合物的实验操作,只改变投料过程的b2碱(磷酸钾)的用量(相对式7的原料摩尔比)。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表12。
表12
实施例18:合成式9化合物
不同溶剂用量对比研究:合成实验组参照实施例5中的式9化合物的实验操作,只改变投料过程的s2溶剂(甲苯)的用量(相对式7的原料投料量)。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表13。
表13
实施例19:合成式7化合物
不同温度t2对比研究:合成实验组参照实施例5中的式9化合物的实验操作(溶剂为dmso),只改变反应过程反应液的t2温度参数。其余的投料参数和反应参数不变。经过投料,反应,处理,精制纯化得出的式9化合物的纯度和收率见表14。
表14
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!