一种检测细胞内基因编辑效率及脱靶的方法与流程
本发明涉及基因分析检测技术领域,尤其涉及一种检测细胞内基因编辑效率及脱靶的方法。
背景技术:
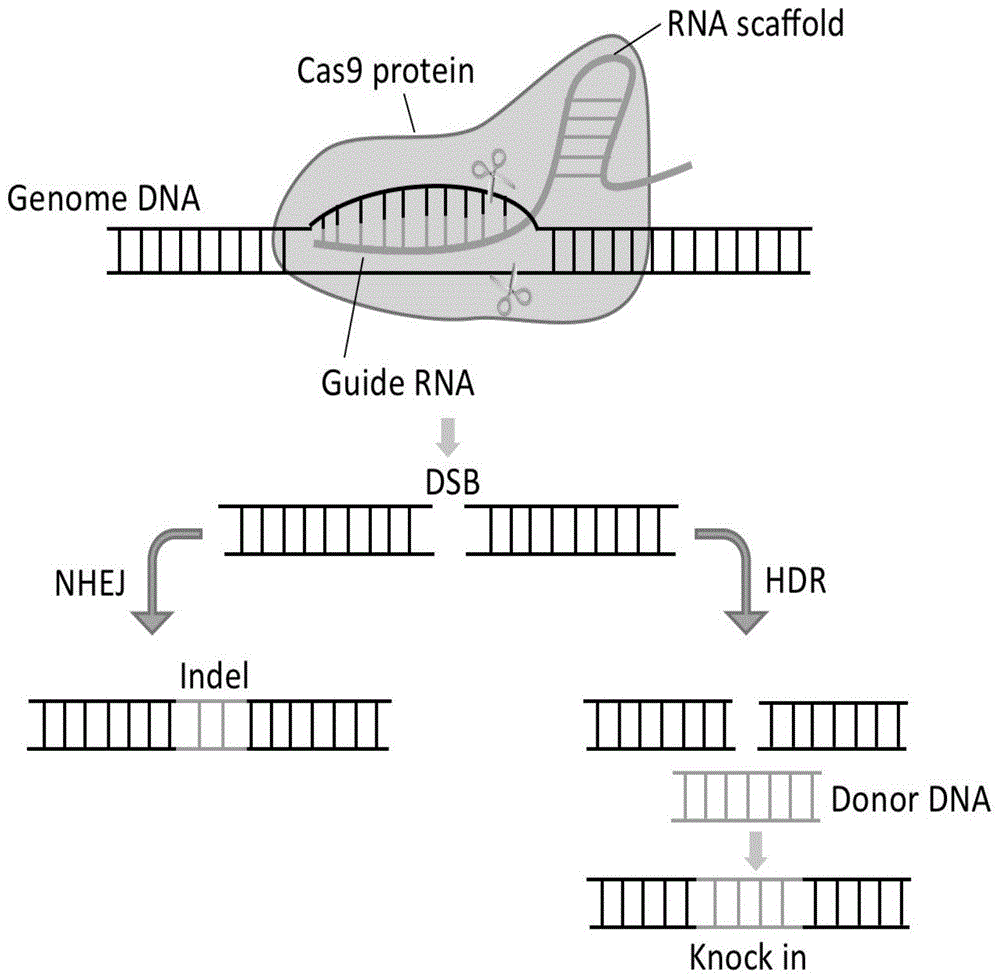
crispr/cas9系统是一种简单高效的基因编辑工具。其由两部分组成,引导rna(guiderna,grna)和cas9蛋白。引导grna可指导cas9蛋白结合到目的基因片段发挥核酸内切酶的作用对靶片段(dna双链)进行剪切造成双链断裂(doublestrandbreak,dsb),机体在对损伤的dna区域进行修复时会随机引入碱基突变,有一定的概率造成基因突变;如果在修复时另外提供一个同源修复的模板dna,则可对靶基因区域进行定向改造,其原理如图1所示。
crispr/cas9系统具有具有操作简单、价格低廉、效率高等优点,但也会存在一定的脱靶现象。crispr的脱靶现象会增加疾病治疗的风险,阻碍临床的进一步应用,所以提高crispr/cas9精确性和解决crispr/cas脱靶问题是基因编辑领域的重中之重。目前主要是通过寻找新的crispr/cas系统和进化改造现有的cas蛋白。但是如何使用简单高效的方法高通量检测crispr系统基因编辑器的编辑效率、编辑图谱和脱靶效应,无疑是现在亟待解决的一个问题。
crispr/cas系统只需要一个靶向目标序列的grna就能引导cas相关核酸酶结合相应的基因序列,并指导切割该位点。为了提高cas9的特异性,科学家除了寻找cas9同系物外,还通过cas9改造来提高crispr/cas9系统的特异性,该策略主要是通过基因工程手段使cas9的双链切割突变为单链切割。这样只有2条sgrna同时发生错配时才会发生脱靶,这就会极大降低脱靶效应。cas9蛋白都需要grna引导才能切割dna,因此通过调整grna来提升特异性就适用更多情况,有研究表明截短的grna(小于20bp)和发卡结构的grna能使编辑效率提高且大幅度降低脱靶率。但是如何高通量检测这些方法的编辑效率和脱靶效应是一个非常重要的问题。
现有检测基因编辑效率和脱靶效应的技术主要包括以下几种:细胞外检测方法,例如digenome-seq,circle-seq,site-seq等,这些技术都是在体外用crispr/cas9切割基因组dna,然后高通量测序分析基因编辑效率和脱靶情况,显然这种脱离细胞本身的检测方法难以预测真实细胞内发生的编辑现象。细胞内检测方法,例如guide-seq,discover-seq,goti等,与细胞外检测技术相比,这些方法能很好地模拟细胞内的脱靶现象。guide-seq技术需要一种短双链寡核苷酸来标记crispr-cas诱导的脱靶断裂,然后高通量检测标签所在的基因组区域预测脱靶位置,但该方法不能检测基因脱靶的详细情况,也不能用来检测基因编辑图谱。discover-seq技术是一种利用dna修复因子同时结合染色质免疫共沉淀和高通量测序的方法。goti技术利用双细胞胚胎注射grna和核酸酶来评估核酸酶的脱靶效应。这两种方法虽然提高了检测的准确性,但操作相当复杂,限制了其在基础研究的应用。
总而言之,现有检测基因编辑效率和脱靶效应的技术的缺陷包括:(1)测序成本相对较高,需要全基因组测序,浪费测序资源;(2)通量不足,无法同时检测上千条grna的编辑效率和编辑图谱,无法同时检测成百上千条grna的脱靶效率;(3)部分检测技术需要较复杂精细的操作过程,如discover-seq需要免疫共沉淀;goti需要胚胎受精卵注射及全基因组测序等。
技术实现要素:
本发明提供一种检测细胞内基因编辑效率及脱靶的方法,能够高通量检测基因编辑效率及脱靶,成本较低。
本发明通过以下技术方案实现:
一种检测细胞内基因编辑效率及脱靶的方法,包括:
构建trap-seq序列,其包括从5’端至3’端依次连接的上游接头序列、grna序列、grna骨架序列(scaffold)、grna靶向序列和下游接头序列,其中上述grna靶向序列与上述grna序列互补配对;
使用设计在上述上游接头序列和下游接头序列上的引物,对上述trap-seq序列进行pcr扩增得到双链trap-seq序列;
将上述双链trap-seq序列连接到质粒骨架上,得到重组trap-seq序列质粒库;
对上述重组trap-seq序列质粒库进行慢病毒包装,并感染宿主细胞株;
从被感染的宿主细胞株中提取细胞基因组,然后使用设计在上述质粒骨架上的引物扩增上述细胞基因组获得上述trap-seq序列,然后测序并分析测序数据得到基因编辑效率及脱靶情况。
在优选实施例中,上述目标基因是与人类药靶相关的基因。
在优选实施例中,上述grna靶向序列包括3bppam序列、20bpgrna互补序列以及其上游10bp和下游4bp的基因组序列。
在优选实施例中,上述设计在上述上游接头序列和下游接头序列上的引物的序列分别如下:
trap-oligo(bsmbigga)-f:tacagctaccacgtctcacacc(seqidno:1);
trap-oligo(bsmbigga)-r:agcacaaccgtcgtctccaaac(seqidno:2)。
在优选实施例中,上述质粒骨架包括用于细胞筛选的报告基因。
在优选实施例中,上述报告基因是绿色荧光蛋白报告基因。
在优选实施例中,上述质粒骨架是lenti-trapseq-gfp-puro骨架。
在优选实施例中,上述宿主细胞株是hek293t细胞。
在优选实施例中,上述hek293t细胞包括野生型hek293t细胞、稳定表达spcas9的hek293t细胞和稳定表达abe7.10的hek293t细胞。
在优选实施例中,上述分析测序数据得到基因编辑效率及脱靶情况包括分析spcas9和/或腺嘌呤碱基编辑(abes)的基因编辑图谱和基因编辑效率。
本发明的有益效果包括:
(1)本发明仅通过一对引物就能高通量检测基因编辑效率及脱靶,节约时间和人力;
(2)本发明只需扩增子测序即可获得所需数据量,避免了全基因组测序带来的资源浪费,节约测序成本;
(3)本发明通过常规细胞实验室的配置即可完成整个实验过程,且trap-seq的整个系统中含有荧光报告基因方便筛选和实时观察,因此具有操作简单方便的优势。
附图说明
图1为本发明实施例中crispr/cas9系统基因编辑示意图,crispr/cas9系统由grna和cas9蛋白两部分组成,cas9蛋白可结合grna的二级结构并由grna靶向至基因敲除位点,精准地对靶位点进行切割,后续机体通过nhej和hdr两种方式对损伤dna进行修复。
图2为本发明实施例中trap系统的整个工作流程示意图。
图3为本发明实施例中trapseq设计策略原理图。
图4为本发明实施例中不同扩增循环数的pcr产物核酸电泳结果图。
图5为本发明实施例中trapseq12k芯片(包含12000条grna)在不同水平的覆盖度,从上到下分别为pcr产物水平、质粒库水平和细胞水平。
图6为本发明实施例中lenti-trapseq质粒库构建的蓝白斑筛选结果图。
图7为本发明实施例中慢病毒包装后的滴度检测(流式细胞检测分析)结果图。
图8为本发明实施例中trap-seq的编辑结果与其内源基因编辑结果对比图。
图9为本发明实施例中spcas9在不同时间点的编辑效率结果图。
图10a和图10b为本发明实施例中spcas9在不同时间点的编辑图谱。
图11为本发明实施例中abe7.10的编辑效率和编辑窗口分析结果图。
图12为本发明实施例中abe7.10在编辑位点侧翼的碱基偏好性分析结果图。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。在以下的实施方式中,很多细节描述是为了使得本发明能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他材料、方法所替代。
另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
本发明的检测细胞内基因编辑效率及脱靶的方法,通过体外合成trap-seq序列实现高通量检测crispr的编辑效率和脱靶效应,简单可靠;只需要通过扩增子测序即可获得高通量测序的样本,节约测序资源,成本大大降低;并且,除了能检测crispr的编辑效率、编辑图谱和脱靶图谱外,还能用来筛选药靶基因和小分子药物,应用范围广。
本发明实施例中,一种检测细胞内基因编辑效率及脱靶的方法,如图2所示,包括:
(1)构建trap-seq序列,其包括从5’端至3’端依次连接的上游接头序列、grna序列、grna骨架序列(scaffold)、grna靶向序列和下游接头序列,其中grna靶向序列与grna序列互补配对。
如图3所示,本发明的一个实施例中,上游接头序列是上游bsmbi接头(linker),该接头序列上包括一个bsmbi酶切位点,该接头序列的长度可以是15bp;grna的长度可以是21bp;grna骨架序列(scaffold)的长度是82bp;grna靶向序列包括3bppam序列、20bpgrna互补序列(即与grna序列互补的序列)以及其上游10bp和下游4bp的基因组序列;下游接头序列是下游bsmbi接头(linker),该接头序列上包括一个bsmbi酶切位点,该接头序列的长度可以是15bp。如图3所示,trap-seq序列被连接入一个载体中(即lenti-trapseq-gfp-puro骨架),将grna靶向序列(37bp)命名为trap,而将从上游bsmbi接头到下游bsmbi接头在内的所有序列称为trap-seq。
(2)使用设计在上述上游接头序列和下游接头序列上的引物,对上述trap-seq序列进行pcr扩增得到双链trap-seq序列,即通过pcr方法将trap-seq文库的寡核苷酸(oligos)变为dsdna,命名为trap-crispr-pcrpool(池)。
在本发明的一个实施例中,上游接头序列和下游接头序列分别是上游bsmbi接头(linker)和下游bsmbi接头(linker),相应地,设计在上游接头序列和下游接头序列上的引物分别是:
trap-oligo(bsmbigga)-f:tacagctaccacgtctcacacc(seqidno:1);
trap-oligo(bsmbigga)-r:agcacaaccgtcgtctccaaac(seqidno:2)。
(3)将上述双链trap-seq序列连接到质粒骨架上,得到重组trap-seq序列质粒库。
在本发明的一个实施例中,质粒骨架包括用于细胞筛选的报告基因,例如绿色荧光蛋白报告基因,特别是,如图3所示,以lenti-trapseq-gfp-puro作为质粒骨架。
在本发明的一个实施例中,以lenti-trapseq-gfp-puro作为质粒骨架,实现lenti-trapseq-质粒库的构建。具体而言,trap-crispr-pcrpool(池)通过golden-gate方法与lenti-trapseq-gfp-puro骨架连接,此质粒骨架中包括绿色荧光蛋白报告基因,便于后期的细胞筛选。
(4)对上述重组trap-seq序列质粒库进行慢病毒包装,并感染宿主细胞株,例如hek293t细胞,特别是,野生型hek293t细胞、稳定表达spcas9的hek293t细胞和稳定表达abe7.10的hek293t细胞等。
(5)从被感染的宿主细胞株中提取细胞基因组,然后使用设计在上述质粒骨架上的引物扩增上述细胞基因组获得上述trap-seq序列,然后测序并分析测序数据得到基因编辑效率及脱靶情况,例如,分析spcas9和/或腺嘌呤碱基编辑(abes)的基因编辑图谱和基因编辑效率。
慢病毒感染宿主细胞株后,grna靶向的目的基因(trap)将会发生编辑,在trap-seq上下游的lenti-trapseq-gfp-puro骨架上设计引物,通过pcr方法扩增出trap-seq序列,然后ngs高通量测序及数据分析。
在本发明的一个实施例中,在trap-seq上下游的lenti-trapseq-gfp-puro骨架上设计的引物序列如下:
trapseq-ngs-f1:ggactatcatatgcttaccgta(seqidno:3);
trapseq-ngs-r1:actcctttcaagacctagctag(seqidno:4)。
以下通过实施例详细说明本发明的技术方案和效果,应当理解,实施例仅是示例性的,不能理解为对本发明的限制。
实施例1
本实施例中,通过trap-seq该方法分别检测spcas9和腺嘌呤碱基编辑(abe7.10)的编辑效率和编辑图谱。
1.grna的选择。
trap-seq(targetedreporter-anchoredpositionalsequencing)方法设计策略是:将上游bsmbilinker(15bp),grna(21bp),grnascaffold(82bp),grna靶向序列(3bppam序列、20bpgrna序列和其上游10bp、下游4bp的基因组序列)和下游bsmbilinker(15bp)放在同一个载体(lenti-trapseq-gfp-puro骨架)中,将grna靶向序列(37bp)命名为trap(图3),而将从上游bsmbilinker到下游bsmbilinker在内的所有序列称为trap-seq。
选择与人类药靶相关的7595个基因设计grna,输出所有的grna,然后过滤掉一些潜在脱靶较多的grna(flashfryversion1.80,https://github.com/mckennalab/flashfry),输出12000条trap-seq序列信息,最后移交基因合成公司合成trap-seq文库。
2.将trap-seq文库的寡核苷酸(trapseq-oligos)变为dsdna,实施过程如下:
将trap-seq文库的寡核苷酸(trapseq-oligos)稀释到1ng/μl作为模板,进行如下pcr扩增。
pcr体系如下表1所示:
表1
引物序列为:
trap-oligo(bsmbigga)-f:tacagctaccacgtctcacacc(seqidno:1);
trap-oligo(bsmbigga)-r:agcacaaccgtcgtctccaaac(seqidno:2)。
该引物的上游加上了(tacagct)、下游加上了(agcacaa)平衡碱基,以实现引物序列的稳定。
温控反应程序为:98℃2min,(98℃10s,55℃10s,72℃30s)×不同的循环数,72℃7min,最终4℃保温。
实验过程中发现不同的pcr循环数扩增的条带大小不同,根据上述pcr体系探索最佳pcr扩增循环数,发现在21个循环时能扩增出大概184bp条带,符合目的条带长度(图4)。所以实验采用21个循环数扩增。
上述pcr体系扩增36管,过柱回收的pcr产物命名为trap-crispr-pcrpool(池),其中一部分用来ngs质控(图5),剩下用来构建lenti-trapseq质粒库。
3.lenti-trapseq质粒库的构建
本实施例中骨架采用lenti-trapseq-gfp-puro。
3.1golden-gate的连接
连接体系如下表2所示:
表2
温控反应程序为:37℃5min,10个循环;22℃10min;37℃30min;75℃15min;4℃保温。
3.2连接产物的转化
具体步骤如下:
a.准备以下表3所示的实验材料,于冰上放置3min。
表3
b.加入25μl感受态细胞(dh5α)到步骤a的产物中,冰上放置20min,室温放置10min,然后加入150μl经过37℃预热的lb培养基,放37℃摇床1h后,取200μl均匀涂布到lb平板上(该平板含有x-gal+iptg+amp),37℃培养箱倒置培养16h(±2h)。预实验发现上述体系转化的平板长出的菌落密度和蓝白板比例合适,如图6所示,本实例共转化36个15cmlb平板,以确保lenti-trapseq质粒库的覆盖度。
c.提取lenti-trapseq质粒库,收集步骤b中全部菌落,用无内毒素质粒试剂盒(invitrogen,endotoxin-freemaxiplasmidpurificationkit)提取质粒,命名为trapseq-plasmidslibrary(质粒文库),然后在trap-seq上下游的lenti-trapseq-gfp-puro骨架上设计引物,通过pcr方法扩增出trap-seq文库序列。
pcr体系如下表4所示:
表4
引物序列如下:
trapseq-ngs-f1:ggactatcatatgcttaccgta(seqidno:3);
trapseq-ngs-r1:actcctttcaagacctagctag(seqidno:4)。
该引物扩增条带大小为252bp。
温控反应程序为:98℃2min,(98℃10s,55℃10s,72℃30s)×22个循环,72℃7min,4℃保温。
上述pcr体系扩增72管,胶回收pcr产物,ngs测序质控(图5)。
4.lenti-trapseq质粒库的慢病毒包装
4.1病毒包装
本实施例采用最为常用的三质粒慢病毒包装系统(prsv-rev,pmd-lg/p-rre,pmd.2g),当hek293t细胞密度培养至70%时做转染,转染体系如下表5所示:
表5
转染24h后更换新鲜培养基,48h后收集培养基用0.45μm滤膜过滤,滤液中加入8μg/ml的聚凝胺(polybrene)存放-80℃保存,命名为lenti-trapseqviruslibrary(病毒文库),把所有的病毒分装两份,分别命名为lenti-trapseqviruslibrary1(病毒文库1)和lenti-trapseqviruslibrary2(病毒文库2)。
4.2病毒滴度检测
24孔板里hek293t细胞培养至50-70%时感染lenti-trapseqviruslibrary1和lenti-trapseqviruslibrary2,详细的感染体积包括0μl、5μl、10μl、20μl、40μl、80μl、160μl、320μl,每个体积两个重复。
感染后24h换液,48h收集细胞用流式仪检测每组细胞绿色荧光的比例,然后根据病毒感染前细胞的数量计算出病毒滴度,计算方法为:lenti-trapseqviruslibrarytiter=绿色荧光蛋白的比例×病毒感染前细胞数量。每组绿色荧光细胞比例如图7。根据细胞中绿色荧光蛋白的比较可以计算出lenti-trapseqviruslibrary的滴度。
5.lenti-trapseq慢病毒感染hek293t细胞及嘌呤霉素(puromycin)筛选
实验分为五组:野生型hek293t细胞组,稳定表达spcas9的hek293t细胞加dox诱导组(spcas9+dox),稳定表达spcas9的hek293t细胞无dox诱导组(spcas9-dox),稳定表达abe7.10的hek293t细胞加dox诱导组(abe+dox),稳定表达abe7.10的hek293t细胞无dox诱导组(abe7.10-dox)。感染方案如下:根据moi=0.3感染细胞,每组共感染10个10cm皿,感染24h后换液,48h后加嘌呤霉素(puromycin)连续筛选9天,选不同时间点收集部分细胞并用荧光显微镜记录绿色荧光蛋白表达情况。
细胞收集时间点如下表6所示:
表6
注:+表明在此时间点收细胞。
6.细胞基因组提取和trap核心部位扩增
6.1大量提取细胞基因组的方法如下:
a.取1×107细胞,pbs缓冲液清洗后,放入1.5ml或合适的离心管中;
b.向离心管中加入0.45mlsnet溶液(20mmol/ltris-hcl(ph8.0),5mmol/ledta(ph8.0),400mmol/lnacl,1%sds(m/v)),50μlsds(10%),以及5μl蛋白酶k(20mg/ml),充分混匀后,于56℃孵育4-6h或过夜;
c.放置至室温,加入等体积的饱和酚,颠倒混匀,12000rpm离心10min,小心吸取上层清液到新的1.5ml或合适的离心管中;
d.加入等体积的酚:氯仿:异戊醇(25:24:1),颠倒混匀,12000rpm离心10min,取上层清液到新的1.5ml合适的离心管中;
e.加入等体积氯仿:异戊醇(24:1),颠倒混匀,12000rpm离心10min,取上层清液到新的1.5ml合适的离心管中;
f.加入2.5倍体积的-20℃预冷的无水乙醇,轻摇混匀,观察析出的絮状沉淀;
g.12000rpm离心10min,弃乙醇;
h.加入500ul的-20℃预冷的75%乙醇,10000rpm离心5min,弃乙醇,超净台中将dna吹干;
i.根据dna的多少,加入适量超纯水溶解dna(一般为50-200μl),加入适量(一般2-5μl)rnasea(20mg/ml)37℃消化30min;
j.取1μl样品测定样品浓度以及纯度,260/280nm测od值在1.8~2.0之间,260/230nm测od值在2.0~3.0之间为佳,并取约100ng样品电泳检测其是否降解;样品于-20℃保存备用。
6.2trap核心部位pcr扩增及ngs测序
pcr体系如下表7所示:
表7
温控反应程序为:94℃2min,(94℃20s,58℃30s,68℃45s)×25循环,68℃7min,4℃保温。
每个实验组按照上述pcr体系扩增24管,胶回收后通过pcr-free建库的方式进行ngs测序。
7.数据分析
7.1trap-seq的编辑结果与其内源基因编辑结果对比
开始trap-seq建库实验前,我们首先用单个trap-seq质粒(靶向aavs1质粒)来测试该方法的基因编辑结果和内源性基因编辑结果是否有差异。通过pcr分别扩增出trap-seq序列和内源性基因序列,然后sanger测序后,使用tide(https://tide.deskgen.com/)分析基因编辑结果,如图8所示,发现aavs1trap的编辑效率和aavs1基因组的编辑效率都能达到80%以上,且基因的编辑类型(缺失或插入)相似,说明trap-seq的方法能反应出细胞内源基因编辑效率。
7.2spcas9在不同时间点产生indel频率变化
为了评估测序质量并过滤低质量的测序数据,我们使用fastqc-0.11.3(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)和fastp-0.19.6(https://github.com/opengene/fastp)进行过滤并输出每个样品数据集。所有分析结果均基于过滤后数据,使用python-3.6和r语言来计算整个过程。对编辑效率我们采用(indel读长计数)/(所有净读长计数)的公式来量化,通过分析wt组和spcas9组在不同时间点产生indel的读长(reads)数,发现随着筛选时间的增加indel的频率也在增加(图9),spcs9偏好于1个插入(insertions)的编辑(图10a)和1个缺失(deletions)的编辑(图10b)。由于spcas9的碱基编辑作用,spcas9组的细胞有富集和消减现象。
7.3腺嘌呤碱基编辑器(abe7.10)的编辑窗口
abe是一种腺嘌呤碱基编辑器,abe能将dna中的a.t碱基对转换为g.c碱基对(a>g),从而允许修复某些细胞类型中的突变。本实例中我们用trap-seq系统进一步检测abe7.10的编辑效率和编辑窗口。对abe的编辑效率我们采用(snp读长计数(a>g))/(所有净读长计数)公式来量化,python-3.6和r语言实现数据分析,如图11a,表明abe7.10在1-5%的编辑效率的grna最多,平均编辑效率为20%,编辑窗口为9bp,其中的6个为热点编辑窗口(图11b)。在所有发生编辑碱基替换(a>g)的grna中,我们分析了编辑位点a侧翼的碱基偏好性,发现在a左侧偏好t/g,右侧偏好c,左右两侧排斥a,即当基因上a碱基的左侧为t/g,右侧为c时,该a碱基突变为g碱基的效率较高,而在a碱基左右都为a,那该a碱基突变为g的效率不高,如图12。该发现为对以后grna的选择提供指导意义。
本实施例中我们用trap系统分析了spcas9和腺嘌呤碱基编辑(abes)的基因编辑效率图谱,结果显示该系统能很好地在基因组范围内检测基因编辑图谱和基因编辑效率。
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
sequencelisting
<110>青岛华大基因研究院
<120>一种检测细胞内基因编辑效率及脱靶的方法
<130>19i29238
<160>4
<170>patentinversion3.3
<210>1
<211>22
<212>dna
<213>人工序列
<400>1
tacagctaccacgtctcacacc22
<210>2
<211>22
<212>dna
<213>人工序列
<400>2
agcacaaccgtcgtctccaaac22
<210>3
<211>22
<212>dna
<213>人工序列
<400>3
ggactatcatatgcttaccgta22
<210>4
<211>22
<212>dna
<213>人工序列
<400>4
actcctttcaagacctagctag22
- 还没有人留言评论。精彩留言会获得点赞!