一种高纯度罗库溴铵的制备方法与流程
本发明涉及医药领域,具体涉及罗库溴铵的制备。
背景技术:
肌肉松弛药是外科手术必须的麻醉辅助药,用量很大,目前常见的有罗库溴铵、维库溴铵、琥珀胆碱、阿曲库铵等。rocuroniumbromide(中文名:罗库溴铵),化学名为1-[(2β,3α,5α,16β,17β)-17-(乙酰氧基)-3-羟基-2-(4-吗啉基)雄甾烷-16-基]-1-(2-丙烯基)吡咯烷溴化物,是荷兰organon公司开发的甾类非去极化肌肉松弛剂。由于罗库溴铵起效最快,无释放组胺等副作用,使之成为同类药中最有前途的药物。自荷兰欧加农公司开发以来,先后在荷兰、比利时、德国、美国、英国、日本、香港等四十多个国家和地区上市销售,并获得广泛认可及好评,1998年在世界500种处方药物销售额处于领先之位。结构式如式ⅰ所示:
关于罗库溴铵制备工艺的文献较多,如专利us2006/0058275、wo2007/03348等,报道的合成路线基本一致,均是采用3α,17β-(二羟基)-2β-(吗啉-1-基)-16-(吡咯烷-1-基)-5α-雄甾烷为原料,首先乙酰化生成中间体(2β,3α,5α,16β,17β)-17-乙酰氧基-3-羟基-2-(4-吗啉基)-16-(1-吡咯烷基)雄甾烷,再与3-溴丙烯成季铵盐生成罗库溴铵,其合成路线如下:
通过文献检索发现,很多文献针对罗库溴铵的反应过程进行了报道,但报道罗库溴铵纯化方法的文献较少。罗库溴铵本身对湿、热和氧较为敏感,极易发生降解,不易纯化、干燥和储运,难以按照常规的精制方法进行纯化,罗库溴铵的纯化存在以下的难点:
(1)罗库溴铵结构中含有酯基,酯基在湿、热、氧化等条件下,较易发生降解。而罗库溴铵为季铵盐,具有引湿性,在反应后处理过程中不可避免会吸湿发生水解生成杂质c。杂质c与罗库溴铵结构相似,通过将低纯度罗库溴铵溶解于良溶剂中,再加入到不良溶剂中析出的常规纯化方法去除效果较差。
(2)罗库溴铵在热、光照条件下,易降解生成杂质a。而罗库溴铵易与有机溶剂形成溶剂合物,干燥去除困难,在长时间加热干燥过程中,会导致杂质a增大。而通过冻干法进行干燥,会人为引入水分,导致杂质c的增大。
(3)在罗库溴铵反应过程中,产生的小杂质较多,性质与罗库溴铵相似。通过常规纯化方法去除困难。
罗库溴铵降解产生杂质a和杂质c路线如下;
由于上述情况的综合作用,使得罗库溴铵在生产、纯化、干燥等生产环节,会引入较多杂质,纯化难度非常大。在罗库溴铵的制备过程中,一般情况下,通过反应制备的低纯度罗库溴铵总杂可以达到2.0%~1.0%。经一般方法纯化后可以达到总杂1.0%~0.5%,进一步地可纯化至总杂在0.5%~0.2%。然而,想进一步获取更高纯度的罗库溴铵,需要损失大量产品,另外,引入的有机溶剂会与罗库溴铵形成溶剂合物,较难去除。同时由于罗库溴铵本身的不稳定性,导致罗库溴铵的纯化是其生产工艺中难度最大的环节。
cn101993470a公开了一种制备高纯度罗库溴铵的工艺,将低沸点反溶剂和含有机溶剂或/和杂质的罗库溴铵混合,利用有机溶剂或/和杂质溶于低沸点反溶剂,而罗库溴铵不溶于或难溶于低沸点反溶剂的原理带走罗库溴铵中的有机溶剂或/和杂质,使罗库溴铵沉淀或结晶析出,进而得到不含有机溶剂或/和杂质的高纯度罗库溴铵,这种方法对设备要求较高,应用高压设备,不适合工业化操作,且存在一定安全风险;cn103435674公开了一种高纯度、高稳定性罗库溴铵的制备方法,先将罗库溴铵粗品用氧化铝预处理除去大部分3-溴丙烯,过滤,将所得滤液滴加到剧烈搅拌的乙醚中,析出大量白色固体,再将得到的白色固体溶于稀醋酸溶液中,将该溶液快速冻结成冰,经冷冻干燥得到罗库溴铵。该专利采用大量的乙醚、冻干等手段进行纯化,安全风险较大,同时设备投入也较高;cn108570090a公开了一种高纯度罗库溴铵的制备方法,将罗库溴铵粗品溶于丙酮水溶液、过滤、减压浓缩,加入正庚烷均质、打浆等步骤得到高纯度罗库溴铵,该方法需要减压浓缩丙酮水溶液,使产品存在较大的质量风险,且纯化过程操作复杂。这些公开的现有技术一方面制造工艺复杂且设备昂贵,另一方面制得的罗库溴铵,杂质总量一般都在0.2%以上。
因此,开发一种在生产上易于实现的、最大程度降低纯化过程中产品分解的、安全风险高的纯化方法是在罗库溴铵的工艺和质量研究过程中急切需要解决的问题。
技术实现要素:
基于上述问题,本发明提供一种高纯度罗库溴铵的制备方法,该制备方法解决了罗库溴铵纯化过程中易降解、吸潮、易形成溶剂合物不易干燥等问题,可提高罗库溴铵的纯度至99.95%以上。
技术方案是:一种高纯度罗库溴铵的制备方法,该方法通过将低纯度罗库溴铵在有机溶剂中重结晶而制取高纯度罗库溴铵。本发明中重结晶是指将低纯度罗库溴铵溶解于溶剂中形成均相溶液后再结晶析出。
作为优选,所述有机溶剂为有机溶剂a或有机溶剂a与有机溶剂b的混合物;
所述有机溶剂a选自丙酮、丁酮、甲基异丙基甲酮、二氯甲烷中的一种或多种,有机溶剂b选自甲醇、乙醇、四氢呋喃、甲基叔丁基醚、乙醚、乙酸乙酯、乙酸异丙酯、异丙醇、乙腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺中的一种或多种;优选有机溶剂a选自丙酮、丁酮中的一种或二种,所述有机溶剂b选自乙腈、四氢呋喃、甲醇、乙醇、异丙醇、甲基叔丁基醚、乙醚中的一种或多种。
作为优选,所述有机溶剂a选自丙酮、丁酮中的一种或二种,所述有机溶剂b选自乙腈、四氢呋喃、甲醇、乙醇、异丙醇、甲基叔丁基醚、乙醚中的一种或多种。
作为优选,所述有机混合溶剂重量为低纯度罗库溴铵重量的0.5~50倍,优选2~10倍。
技术方案是:一种罗库溴铵的制备方法,该方法包括以下步骤:
①低纯度罗库溴铵加入到有机溶剂中,搅拌使固体溶解;
②析晶,过滤;
③干燥得高纯度罗库溴铵。
该制备方法制备出的高纯度罗库溴铵,稳定性较好,能够解决或者减缓受热与遇水降解的风险,可应用于罗库溴铵生产、储存和运输。
作为优选,所述有机溶剂为有机溶剂a或有机溶剂a与有机溶剂b的混合物;
所述有机溶剂a选自丙酮、丁酮、甲基异丙基甲酮、二氯甲烷中的一种或多种,有机溶剂b选自甲醇、乙醇、四氢呋喃、甲基叔丁基醚、乙醚、乙酸乙酯、异丙醇、乙酸异丙酯、乙腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺中的一种或多种。
作为优选,所述有机溶剂a选自丙酮、丁酮中的一种或二种,所述有机溶剂b选自乙腈、四氢呋喃、甲醇、乙醇、异丙醇、甲基叔丁基醚、乙醚中的一种或多种。
作为优选,所述有机混合溶剂重量为低纯度罗库溴铵重量的0.5~50倍。
作为优选,所述有机混合溶剂重量为低纯度罗库溴铵重量的2~10倍。
析晶温度需在较低温度下进行析晶,温度越低,收率越高,因此析晶温度在-80~30℃,为便于生产放大,析晶温度在-30~10℃。
析晶时间越长,收率越高,因此析晶时间在3~100小时,为便于生产放大,析晶时间在10~72小时。
由于罗库溴铵热稳定性较差,同时在低温时溶解性差,因此溶解的溶解温度在0~80℃,当溶解的溶解温度在20~60℃可以取得较好的效果。
本发明所制得的高纯度罗库溴铵具有以下特征:
(1)杂质含量低:单一杂质低于0.02%,总杂质低于0.05%;
(2)残留溶剂符合中国药典标准;基因毒性杂质3-溴丙烯低于0.5ppm或未检出;
(3)产品在影响因素试验的高温25℃的苛刻条件下(敞口放置,不带包装;ep标准规定应在-15℃以下储存;usp标准规定无醋酸的情况下,应在密封容器内,避光、防潮,贮藏于-20℃或更低温度下储存)考察30天,有关物质未发生变化。
本发明的原理及与现有技术相比的有益效果在于:
(1)通过重结晶进行纯化是药物提纯的一般方法,适用于大多数原料药。对于罗库溴铵,由于其结构存在不稳定性,受热易分解,因此通过常规重结晶手段进行纯化会导致其杂质增大,未见通过重结晶进行纯化的文献报道。
本发明通过重结晶法去除杂质,生产工艺及操作简单,使用的均为常规溶剂和生产设备,节约能耗,绿色环保,无安全风险;
(2)去除杂质的效果显著,大大降低了产品中的杂质水平,提高了用药安全性;
(3)产品稳定性具有显著优势,经影响因素的高温25℃苛刻条件考察,有关物质未发生变化。降低了该品种对于储存和运输条件的要求,大大节约了储运成本。
(4)产品具有静电性低的特点,有利于原料药和制剂的生产操作。
(5)制备方法符合工厂gmp生产要求,适合工业化生产。
附图说明
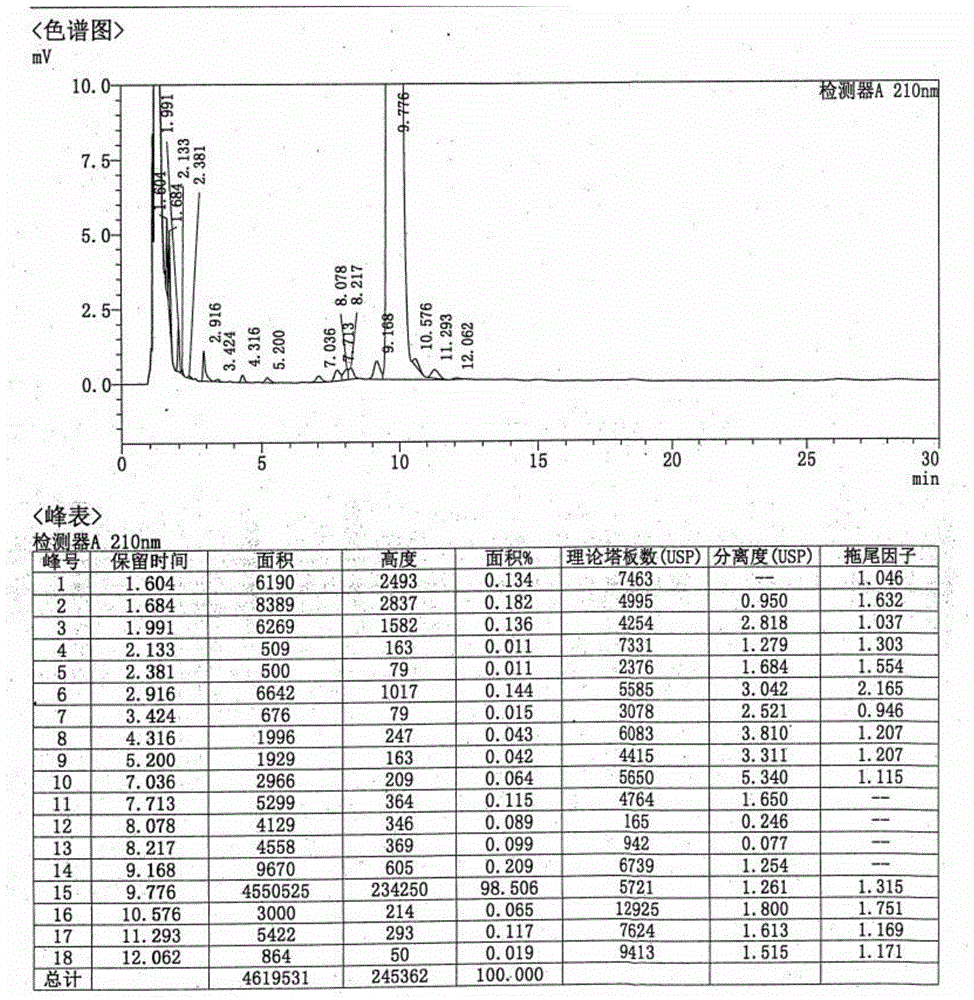
图1是实施例1的低纯度罗库溴铵的hplc图谱;
图2是本发明高纯度罗库溴铵的hplc图谱;
具体实施方式
下面将结合附图对本发明作进一步说明。
本发明提供的实施例仅为进一步解释本发明,不应解释为对本发明的任何限制。
本领域技术人员应当清楚,在下文中,如果未特别说明,本发明所用材料和操作方法是本领域公知的。
本发明所用的检测仪器:
高效液相色谱:岛津lc-2030c。
测试方法为:
照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。用氨基键合硅胶为填充剂的色谱柱(如sepaxhp-amino,4.6×250mm,5μm,
本方法为经国家食品药品监督管理局药品审评中心批准的检测方法。
实施例1:低纯度罗库溴铵的制备
向1000ml烧瓶中加入500g二氯甲烷,加入(2β,3α,5α,16β,17β)-17-乙酰氧基-3-羟基-2-(4-吗啉基)-16-(1-吡咯烷基)雄甾烷100g,搅拌至固体完全溶解,加入100g碳酸钠和100g3-溴丙烯,于20~30℃反应。反应完毕,过滤去除无机盐,二氯甲烷洗涤滤饼,滤液滴入至4000g乙醚中,搅拌分散后,过滤,洗涤,45~50℃减压干燥得罗库溴铵119g,收率95.2%,纯度98.5%。低纯度罗库溴铵hplc图谱如图1。
实施例2:高纯度罗库溴铵的制备
将实施例1制备的10g低纯度罗库溴铵加入到50g丙酮与1g甲醇、2.5g乙醚中,20~30℃搅拌使固体溶解,于-20~-15℃搅拌析晶48小时,过滤,10~20℃减压干燥得高纯度罗库溴铵6.78g,收率67.8%,最大单一杂质0.015%,总杂0.040%。
经检测,其hplc图谱如图2所示。
实施例3:高纯度罗库溴铵的制备
将实施例1制备的11g低纯度罗库溴铵加入到45g丁酮、1.2g甲醇和2.0g甲基叔丁基醚中,20~30℃搅拌使固体溶解,于-10~-5℃搅拌析晶30小时,过滤,10~20℃减压干燥得高纯度罗库溴铵5.76g,收率52.3%,最大单一杂质0.014%,总杂0.034%。
经检测,其hplc图谱与图2基本一致。
实施例4:高纯度罗库溴铵的制备
将实施例1制备的10g低纯度罗库溴铵加入到50g丙酮与1.5g乙醇中,30~35℃搅拌使固体溶解,于-35~-30℃搅拌析晶60小时,过滤,10~20℃减压干燥得高纯度罗库溴铵6.01g,收率60.1%,最大单一杂质0.012%,总杂0.035%。
经检测,其hplc图谱与图2基本一致。
实施例5:高纯度罗库溴铵的制备
将实施例1制备的10g低纯度罗库溴铵加入到55g丙酮与0.8g甲醇、1.7g乙醚中,20~30℃搅拌使固体溶解,于-20~-15℃搅拌析晶48小时,过滤,10~20℃减压干燥得高纯度罗库溴铵6.55g,收率65.5%,最大单一杂质0.014%,总杂0.042%。
经检测,其hplc图谱与图2基本一致。
实施例6:高纯度罗库溴铵的制备
将实施例1制备的14g低纯度罗库溴铵加入到50g丙酮与1g四氢呋喃中,20~30℃搅拌使固体溶解,于-25~-20℃搅拌析晶72小时,过滤,10~20℃鼓风干燥得高纯度罗库溴铵9.18g,收率65.6%,最大单一杂质0.015%,总杂0.044%。
经检测,其hplc图谱与图2基本一致。
实施例7:
将实施例1制备的10g低纯度罗库溴铵加入到550g丙酮中,25~30℃搅拌使固体溶解,于-20~-15℃搅拌析晶36小时,过滤,10~20℃鼓风干燥得高纯度罗库溴铵6.89g,收率68.9%,最大单一杂质0.015%,总杂0.047%。
经检测,其hplc图谱与图2基本一致。
实施例8:静电性试验
将实施例2-7制备的高纯度罗库溴铵分别放入包装袋,剧烈振摇后倒出,未发现有罗库溴铵留在包装袋内表面上。
实施例9:稳定性试验
取实施例2制备的高纯度罗库溴铵进行影响因素试验,考察其稳定性,纯度检测用hplc法。测试方法为:
照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。用氨基键合硅胶为填充剂的色谱柱(如sepaxhp-amino,4.6×250mm,5μm,
本方法为经国家食品药品监督管理局药品审评中心批准的检测方法。
影响因素试验的高温条件下考察30天后经检测,其有关物质未发生变化,其hplc图谱与图2基本一致。稳定性数据如下:
由上述数据可以看出,本发明制备得到的高纯度罗库溴铵的质量水平远远高于各国药典的质量控制要求,且具有良好的化学稳定性,经影响因素高温30天的苛刻条件考察,其有关物质未发生变化,可提升药品在生产后的长期储存和运输过程中的质量水平。同时不需要在欧美药典中规定的苛刻条件下进行储运,可大大降低储运成本。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!