一种蛇毒肽SYN-AKE的液相合成方法与流程
一种蛇毒肽syn-ake的液相合成方法
技术领域
1.本发明属于制药领域,具体涉及一种蛇毒肽syn-ake的液相合成方法。
背景技术:
2.syn-ake为dsm全资子公司pentapharm 2007年开发的一种模拟蛇毒毒素waglerin i活性的类蛇毒三肽。和肉毒杆菌和阿基瑞林一样,在除皱方面亦有特效,但是相比肉毒杆菌却更安全有效。肽序为h-β-ala-pro-dab-nhbzl,分子结构如下所示:
[0003][0004]
文献tetrahedron letters 55(2014)5745
–
5747介绍了一种以不保护氨基酸为原料,最后一步采用霍夫曼重排反应全液相合成syn-ake的方法。专利cn201711270645公开了一种类蛇毒三肽的液相合成方法,该方法将boc-β-ala-oh和h-pro-ome
·
hcl为原料制备boc-β-ala-pro-ome,进一步用lioh处理得到boc-β-ala-pro-oh;采用相同方法和h-dab(boc)-ome
·
hcl反应得到boc-β-ala-pro-dab(boc)-oh;再和苄胺反应得boc-β-ala-pro-dab(boc)-nh-bzl,最后用三氟乙酸脱去boc保护基并制备纯化得纯度大于95%的类蛇毒三肽。
[0005]
文献tetrahedron letters 55(2014)5745方法步骤繁琐,全合成收率低于10%。专利cn201711270645公开方法以h-pro-ome
·
hcl及h-dab(boc)-ome
·
hcl需要lioh强碱处理,不但增加步骤,而且可能会造成消旋杂质产生。因为需用三氟乙酸脱除boc保护基,三氟乙酸对设备腐蚀性较高。另外,三氟乙酸脱除boc保护基后获得的是syn-ake的三氟乙酸盐,需要通过制备色谱柱进行纯化转盐获得syn-ake的醋酸盐,步骤繁琐,成本增加。
[0006]
为了克服上述缺点,本发明提供了一种合成路线简单、环境友好而且不需制备色谱柱纯化转盐就可直接获得syn-ake醋酸盐形式的合成方法,使得生产成本大为降低。
技术实现要素:
[0007]
为了解决上述背景技术中所提出的问题,本发明的目的在于提供一种蛇毒肽syn-ake的液相合成方法。本发明以trt-β-ala-oh,fmoc-dab(trt)-oh为原料采用2+2合成策略经过逐步偶联获得trt-β-ala-pro-dab(trt)-nhbzl,重结晶获得高纯度trt-β-ala-pro-dab(trt)-nhbzl后用醋酸溶液脱除trt,最用乙醚沉淀后直接得到syn-ake的醋酸盐。
[0008]
为了达到上述目的,本发明所采用的技术方案为:一种蛇毒肽syn-ake的液相合成方法,包括以下步骤:
[0009]
1)化合物trt-β-ala-pro-oh的合成;
[0010]
2)化合物fmoc-dab(trt)-nhbzl的合成;
[0011]
3)化合物h-dab(trt)-nhbzl的合成;
[0012]
4)化合物trt-β-ala-pro-dab(trt)-nhbzl的合成;
[0013]
5)蛇毒肽syn-ake:h-β-ala-pro-dab(trt)-nhbzl的合成。
[0014]
进一步地,所述化合物trt-β-ala-pro-oh的合成方法为:以化合物1(trt-β-ala-oh)和honb或化合物1(trt-β-ala-oh)和hosu为原料,在非质子性溶剂存在的条件下,在dic或dcc作用下进行活化反应后,在碱的作用下与h-pro-oh进行偶联反应得到化合物2(trt-β-ala-pro-oh)。
[0015]
进一步地,所述trt-β-ala-oh和honb或trt-β-ala-oh和hosu的摩尔比均为1:1.0~1.2;
[0016]
所述trt-β-ala-oh和h-pro-oh的摩尔比为1:1.0~1.2;
[0017]
所述活化反应所用的活化剂优选为dic;
[0018]
所述活化反应的温度为室温,反应时间为2-4小时,优选为3小时;
[0019]
所述偶联反应所用的碱为dipea、n-甲基吗啉、碳酸氢钠或碳酸氢钾,优选dipea;
[0020]
所述偶联反应的温度为室温,反应时间为1-3小时,优选为2小时;
[0021]
所述非质子性溶剂包括四氢呋喃、二甲基亚砜、二甲基甲酰胺、二恶烷、乙腈、四氯化碳、二硫化碳、苯、甲苯、氯仿、二氯甲烷,优选为二氯甲烷。
[0022]
进一步地,所述化合物fmoc-dab(trt)-nhbzl的合成方法为:以化合物3(fmoc-dab(trt)-oh)为原料,在非质子性溶剂存在的条件下,与苄胺进行偶联反应得到化合物4(fmoc-dab(trt)-nhbzl)。
[0023]
进一步地,所述fmoc-dab(trt)-oh和苄胺的摩尔比为1:1.0~1.2;
[0024]
所述偶联反应的温度为室温,反应时间为2-4小时,优选为3小时;
[0025]
所述非质子性溶剂包括四氢呋喃、二甲基亚砜、二甲基甲酰胺、二恶烷、乙腈、四氯化碳、二硫化碳、苯、甲苯、氯仿、二氯甲烷,优选为二氯甲烷;
[0026]
所述偶联反应所用的偶联剂为dic和化合物a的组合物或dipea和化合物a和化合物b的组合物,其中化合物a为hoat或hobt,化合物b为pyaop、pybop、hatu、hbtu、tbtu或edc,优选为dipea/edc/hobt组合物;更优选地,所述偶联剂组分摩尔比为edc:hobt:dipea=1:1:3。
[0027]
进一步地,所述化合物h-dab(trt)-nhbzl的合成方法为:以化合物4(fmoc-dab(trt)-nhbzl)为原料,在溶剂存在下,用有机碱脱除fmoc保护基得到化合物5(h-dab(trt)-nhbzl)。
[0028]
进一步地,所述脱除fmoc保护基的有机碱为二乙胺、哌啶或dbu有机碱,优选为二乙胺;
[0029]
所述脱保护反应的温度为室温,反应时间为2-4小时,优选为3小时;
[0030]
所述溶剂包括四氢呋喃、二甲基亚砜、二甲基甲酰胺、二恶烷、乙腈、四氯化碳、二硫化碳、苯、甲苯、氯仿、二氯甲烷、甲醇、乙醇、乙酸乙酯、水,优选二氯甲烷。
[0031]
进一步地,所述化合物trt-β-ala-pro-dab(trt)-nhbzl的合成方法为:以中间体化合物trt-β-ala-pro-oh和化合物h-dab(trt)-nhbzl为原料,在非质子性溶剂存在的条件下,进行偶联反应得到化合物6(trt-β-ala-pro-dab(trt)-nhbzl)。
[0032]
进一步地,所述化合物trt-β-ala-pro-oh和化合物h-dab(trt)-nhbzl的摩尔比为1.0~1.5:1;
[0033]
所述偶联反应的温度为室温,反应时间为2-4小时,优选为3小时;
[0034]
所述非质子性溶剂包括四氢呋喃、二甲基亚砜、二甲基甲酰胺、二恶烷、乙腈、四氯化碳、二硫化碳、苯、甲苯、氯仿、二氯甲烷,优选二氯甲烷;
[0035]
所述偶联反应所用的偶联剂为dic和化合物a的组合物或dipea和化合物a和化合物b的组合物,其中化合物a为hoat或hobt,化合物b为pyaop、pybop、hatu、hbtu、tbtu或edc,优选为dipea/edc/hobt组合物;更优选地,偶联试剂组分摩尔比为edc:hobt:diepa=1:1:3;
[0036]
所述化合物trt-β-ala-pro-dab(trt)-nhbzl粗品用乙酸乙酯溶解后,滴加正己烷析晶得到高纯度trt-β-ala-pro-dab(trt)-nhbzl;所述乙酸乙酯的用量与化合物h-dab(trt)-nhbzl的体积质量比为2~4ml:1g;所述正己烷的用量与化合物h-dab(trt)-nhbzl的体积质量比为6~12ml:1g。
[0037]
进一步地,蛇毒肽syn-ake:h-β-ala-pro-dab(trt)-nhbzl的合成方法为:以化合物trt-β-ala-pro-dab(trt)-nhbzl为原料,在醋酸作用下脱trt保护基。
[0038]
进一步地,所述脱trt保护基所用的裂解液为冰醋酸和水,所述裂解液中hoac和h2o体积比为80~99:20~1;
[0039]
所述裂解液用量与化合物物trt-β-ala-pro-dab(trt)-nhbzl的体积质量比为6~8ml:1g;
[0040]
所述脱trt保护基的温度为40℃
±
10℃,裂解时间为1~3小时,优选为2小时;
[0041]
所述脱trt保护基后用乙醚或甲叔醚进行沉淀,离心得到蛇毒肽syn-ake:h-β-ala-pro-dab(trt)-nhbzl,裂解液与沉降试剂的体积比为1:6~10,优选1:8。
[0042]
与现有技术相比:
[0043]
1)以trt-β-ala-oh,fmoc-dab(trt)-oh为原料避免使用lioh处理氨基酸酯的反应,减少反应步骤,降低消旋杂质产生,提高了中间体纯度。
[0044]
2)通过重结晶获得高纯度trt-β-ala-pro-dab(trt)-nhbzl后用醋酸溶液脱除trt,最用乙醚沉淀后直接得到syn-ake的醋酸盐。避免使用强腐蚀性的三氟乙酸作裂解试剂,环境友好;同时避免了制备纯化转盐过程,节约了成本。
附图说明
[0045]
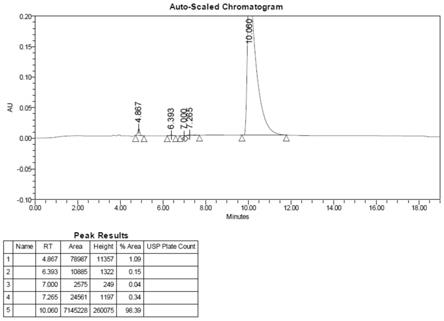
图1为实施例5中syn-ake的hplc谱图;
[0046]
图2为实施例5中syn-ake的质谱图;
[0047]
图3为实施例7中syn-ake的hplc谱图。
具体实施方式
[0048]
为了更好地理解本发明的内容,下面结合具体实施方法对本发明内容作进一步说明,但本发明的保护内容不局限以下实施例。
[0049]
说明书和权利要求书中所使用的缩写的含义列于下表中:
[0050][0051][0052]
syn-ake合成路线如下所示:
[0053][0054]
本发明中的室温为25℃
±
5℃。
[0055]
实施例1:化合物2的合成(trt-β-ala-pro-oh)
[0056]
将trt-β-ala-oh(49.7g,150mmol)、honb(30.0g,165mmol)溶解于350ml dcm中搅拌溶清,冰浴冷却至5℃以下,缓慢加入dic(28ml,180mmol),室温继续搅拌3小时,tlc监控反应。反应完全后,反应液过滤,再加入h-pro-oh(16.0g,180mmol)继续搅拌,滴加入dipea(40.0ml,225mmol)。室温搅拌2小时,tcl监控反应。反应完后,过滤,浓缩。残余物加入350ml乙酸乙酯搅拌溶清,加入饱和柠檬酸溶液,调ph=3~4,静置分层。有机相依次用饱和碳酸氢钠溶液(350ml)以及饱和食盐水溶液(350ml)洗涤,静置分层,有机相用无水硫酸钠干燥过夜。过滤。40℃减压浓缩至100ml。滴加正己烷(400ml),冷却至0℃析晶过夜。过滤,真空干燥得到白色固体60.1g,收率93.4%。
[0057]
实施例2:化合物4的合成(fmoc-dab(trt)-nhbzl)
[0058]
将fmoc-dab(trt)-oh(23.3g,40mmol)、苄胺(5.1g,48mmol)、edc(13.8g,72mmol)和hobt(9.43g,72mmol)加入300ml二氯甲烷中搅拌溶清,冰浴冷却至5℃以下,继续滴加入dipea(37.7ml,216mmol),控制温度不超过10℃。滴加完后室温继续搅拌3小时,tlc监控反应。反应完全后,向反应液中加纯化水(300ml),搅拌15分钟后,静置分液,收集下层有机相。有机相在30℃条件下减压浓缩,残余物用乙酸乙酯(300ml)搅拌溶解,依次用纯化水(300ml)、饱和碳酸氢钠溶液(300ml)、饱和氯化钠溶液(300ml)洗涤,收集有机相,无水硫酸钠干燥过夜。过滤,滤液浓缩至60ml,缓慢滴加入正己烷(240ml)。析出白色固体,继续搅拌1小时后,0℃环境下静置析晶过夜。过滤后40℃真空干燥得到白色固体24.3g,收率90.6%。
[0059]
实施例3:化合物5的合成(h-dab(trt)-nhbzl)
[0060]
将化合物3(24.3g,36.2mmol)加入dcm(250ml)搅拌溶解。将反应液降温至0℃后,缓慢加入二乙胺(26.4g,362.0mol),加完后升至室温继续搅拌3小时。tlc监控反应。原料消耗完后,将反应液浓缩至50ml,再加入正己烷(120ml),冷却至0℃搅拌析出白色固体。真空抽滤,滤饼用正己烷冲洗三次,再将滤饼加入反应瓶内,加正己烷(100ml)打浆2小时。真空抽滤,滤饼在50℃条件下鼓风干燥过夜得到米白色固体15.7g,收率96.3%。
[0061]
实施例4:化合物6的合成(trt-β-ala-pro-dab(trt)-nhbzl)
[0062]
将trt-β-ala-pro-oh(18.0g,42mmol)、h-dab(trt)-nhbzl(15.7g,35.0mmol)、edc(10.0g,52.4mmol)和hobt(7.0g,52.4mmol)加入75ml二氯甲烷中搅拌溶清,冰浴冷却至5℃以下,继续滴加入dipea(27.5ml,157.1mmol),控制温度不超过10℃。滴加完后室温继续搅拌3小时,tlc监控反应。反应完全后,向反应液中加纯化水(75ml),搅拌15分钟后,静置分液,收集下层有机相。有机相在30℃条件下减压浓缩,残余物用乙酸乙酯(100ml)搅拌溶解,依次用纯化水(100ml)、饱和碳酸氢钠溶液(100ml)、饱和氯化钠溶液(100ml)洗涤,收集有机相,无水硫酸钠干燥过夜。过滤,浓缩至50ml,缓慢滴加正己烷150ml。室温析出白色固体,0℃环境下继续静置析晶过夜。过滤后40℃真空干燥得到白色固体27.1g,收率90.1%。
[0063]
实施例5:syn-ake的合成
[0064]
将实施例4中合成的化合物6加入30ml预冻的裂解液(hoac:h2o=95:5)中,室温搅拌3小时。将反应液倒入240ml冰冻乙醚中沉淀,离心,用乙醚洗两次。氮气吹干得到白色固体15.6g。总收率78.8%,hplc纯度98.39%(见图1),ms[m+1]:376(见图2)。
[0065]
实施例6:化合物2的合成(trt-β-ala-pro-oh)
[0066]
将trt-β-ala-oh(24.9g,75mmol)、hosu(9.5g,82.5mmol)溶解于175ml dcm中搅拌溶清,冰浴冷却至5℃以下,缓慢加入dic(14ml,90mmol),室温继续搅拌3小时,tlc监控反
应。反应完全后,反应液过滤,再加入h-pro-oh(8.0g,90mmol)继续搅拌,滴加入dipea(20.0ml,112.5mmol)。室温搅拌2小时,tcl监控反应。反应完后,过滤,浓缩。残余物加入175ml乙酸乙酯搅拌溶清,加入饱和柠檬酸溶液,调ph=3~4,静置分层。有机相依次用饱和碳酸氢钠溶液(175ml)以及饱和食盐水溶液(175ml)洗涤,静置分层,有机相用无水硫酸钠干燥过夜。过滤。40℃减压浓缩至50ml。滴加正己烷(200ml),冷却至0℃析晶过夜。过滤,真空干燥得到白色固体27.1g,收率84.2%。
[0067]
实施例7:syn-ake的合成
[0068]
以实施例6中合成的trt-β-ala-pro-oh为原料,按实施例2~5步骤继续合成获得syn-ake12.1g,总收率61.1%。,hplc纯度99.72%(见图3)。
[0069]
实施例8:syn-ake的合成
[0070]
本实施例步骤1中以trt-β-ala-oh与honb为原料,摩尔比为1:1.0,在室温(25℃
±
5℃)条件下通过活化剂进行活化,活化后在dipea的作用下与h-pro-oh在室温(25℃
±
5℃)进行脱水缩合反应3小时得到化合物2。活化反应选用非质子性溶剂为四氢呋喃。室温(25℃
±
5℃)进行活化反应时间4小时。活化剂为dcc。
[0071]
本实施例步骤2中以fmoc-dab(trt)-oh和苄胺为原料,摩尔比为1:1.0,在室温(25℃
±
5℃)条件下偶联剂进行缩合反应。选用的非质子性溶剂包括四氢呋喃。反应时间4小时,偶联剂为dic和hoat。
[0072]
本实施例步骤3中脱除fmoc的碱为二乙胺,选用的溶剂为四氢呋喃。反应时间2小时。
[0073]
步骤4中以中间体化合物2和化合物5为原料,摩尔比为1:1,在室温(25℃
±
5℃)条件下偶联剂进行缩合反应。选用的非质子性溶剂为四氢呋喃。反应时间2小时,偶联剂为dipea/hobt/pybop。乙酸乙酯的用量与化合物h-dab(trt)-nhbzl的体积质量比(ml/g)为2:1;正己烷的用量与化合物h-dab(trt)-nhbzl的体积质量比(ml/g)为6:1。。
[0074]
步骤5中化合物6为原料,以冰醋酸和水作裂解试剂脱除trt,裂解液hoac/h2o体积比为80:20进行裂解。加热50℃裂解1小时。沉降试剂为甲叔醚,裂解液和沉降试剂的体积比为1:6。氮气吹干得到白色固体14.3g。总收率72.2%,hplc纯度98.04%。
[0075]
实施例9:syn-ake的合成
[0076]
本实施例步骤1中以trt-β-ala-oh与honb为原料,摩尔比为1:1.2,在室温(25℃
±
5℃)条件下通过活化剂进行活化,活化后在dipea的作用下与h-pro-oh在室温(25℃
±
5℃)进行脱水缩合反应1小时得到化合物2。活化反应选用非质子性溶剂为氯仿。室温(25℃
±
5℃)进行活化反应时间2小时。活化偶联剂为dcc。
[0077]
本实施例步骤2中以fmoc-dab(trt)-oh和苄胺为原料,摩尔比为1:1.2,在室温(25℃
±
5℃)条件下偶联剂进行缩合反应。选用的非质子性溶剂为氯仿。反应时间2小时,偶联剂为dipea/hobt/pybop。
[0078]
本实施例步骤3中脱除fmoc的碱为二乙胺,选用的溶剂为氯仿。反应时间4小时。
[0079]
步骤4中以中间体化合物2和化合物5为原料,摩尔比为1.5:1,在室温(25℃
±
5℃)条件下偶联剂进行缩合反应。选用的非质子性溶剂为四氢呋喃。反应时间4小时,偶联剂为dipea/hoat/pyaop。乙酸乙酯的用量与化合物h-dab(trt)-nhbzl的体积质量比(ml/g)为4:1;正己烷的用量与化合物h-dab(trt)-nhbzl的体积质量比(ml/g)为12:1。
[0080]
步骤5中化合物6为原料,以冰醋酸和水作裂解试剂脱除trt,裂解液hoac/h2o体积比为99:1进行裂解。加热30℃裂解3小时。沉降试剂为甲叔醚,裂解液和沉降试剂的体积比为1:10。氮气吹干得到白色固体15.3g。总收率77.3%,hplc纯度98.27%。
[0081]
对比实施例:syn-ake的合成
[0082]
专利cn201711270645公开方法三个实施例中,实施例1总收率最高为38.7%。纯度98.2%,收率明显低于实施例5。
[0083]
以上所述仅为本发明的具体实施方式,不是全部的实施方式,本领域普通技术人员通过阅读本发明说明书而对本发明技术方案采取的任何等效的变换,均为本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1