多氮丙啶化合物的制作方法
多氮丙啶化合物
1.本发明涉及具有至少两个氮丙啶基(或吖丙啶基)的化合物,所述化合物可例如用于使例如溶解和/或分散在水性介质中的羧酸官能聚合物交联。
2.多年来,对具有改善的抗性(如耐沾污性和耐溶剂性)、改善的机械特性和改善的粘合强度的涂料的需求日益增长。这些特性中的一种或多种特性可以借助交联而被提高到更高的水平。多年来已经研究出了许多交联机制,并且对于水性分散体,最有用的交联机制包括羟基官能分散体的异氰酸酯交联、碳二亚胺与羧酸之间的反应、环氧交联以及使用基于氮丙啶的交联剂的交联。
3.us-a-5133997描述了涂料组合物,所述涂料组合物包含直链脂肪族氨基甲酸酯树脂的水性分散体、阴离子表面活性剂,以及能够促进所述树脂固化的交联剂。三羟甲基丙烷三(2-甲基-1-氮丙啶丙酸酯),cas编号64265-57-2,为一种多官能氮丙啶交联剂,被用作交联剂,其为众所周知的并且对于使羧酸官能聚合物交联具有很高的活性。然而,该交联剂具有不利的遗传毒性特征。行业上需要改善粘合剂、油墨和涂料以及用于制备粘合剂、油墨和涂料的物质的安全性、健康和环境特征。遗传毒性描述了导致任何类型的dna损伤的化学或物理试剂的特性,所述dna损伤可能并不总是导致可传递的突变。诱变性是指诱导永久性可传递的dna变化(作为dna组成物或染色体结构),所述变化在体细胞分裂中保持并在生殖细胞中传代到子代。遗传毒性不得与诱变性混淆。所有诱变剂均为遗传毒性的,而并非所有遗传毒性物质均为诱变性的。
4.本发明的目的是提供一种具有至少两个氮丙啶基的化合物,所述化合物与三羟甲基丙烷三(2-甲基-1-氮丙啶丙酸酯)相比具有降低的遗传毒性。具有至少两个氮丙啶基的化合物在本文中还称为多氮丙啶化合物。
5.令人惊讶地,该目的已经通过提供多氮丙啶化合物而得到实现,所述多氮丙啶化合物具有:
6.a)2至6个的以下结构单元(a):
[0007][0008]
其中
[0009]
r1为h;
[0010]
r2和r4独立地选自h、在链中含有1至8个碳原子并且可选地含有一个或多个杂原子的直链基团、在链中含有3至8个碳原子并且可选地含有一个或多个杂原子的支链或环状基团、苯基、苄基或吡啶基;
[0011]
r3为在链中含有1至8个碳原子并且可选地含有一个或多个杂原子的直链基团、在链中含有3至8个碳原子并且可选地含有一个或多个杂原子的支链或环状基团、苯基、苄基或吡啶基;
[0012]
其中r2和r3(在r2与h不同的情况下)可为含有3至8个碳原子的同一环状基团的一
部分;
[0013]
r'=h或含有1至12个碳原子的脂肪族烃基;
[0014]
r
″
=h、含有1至12个碳原子的脂肪族烃基、含有5至12个碳原子的脂环族烃基、含有6至12个碳原子的芳族烃基、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(ocr
″″′
hcr
″″′
h)
n-or
″″″
,其中r
″′
为含有1至12个碳原子的脂肪族烃基并且r
″″
为含有1至12个碳原子的脂肪族烃基或含有6至12个碳原子的芳族烃基,n为1至35,r
″″′
独立地为h或含有1至12个碳原子的脂肪族烃基,并且r
″″″
为含有1至4个碳原子的脂肪族烃基;
[0015]
其中r'和r
″
可为含有5至8个碳原子的同一饱和脂环族烃基的一部分;
[0016]
m为1至6的整数;
[0017]
b)一个或多个连接链(linking chain),其中这些连接链中的每一个连接链连接所述多氮丙啶化合物中存在的所述结构单元a中的两个结构单元a;以及
[0018]
c)在600道尔顿至5000道尔顿范围内的分子量。
[0019]
已经令人惊讶地发现,根据本发明的多氮丙啶化合物与三羟甲基丙烷三(2-甲基-1-氮丙啶丙酸酯)相比具有降低的遗传毒性。根据本发明的多氮丙啶化合物显示出要么仅仅弱阳性诱导的遗传毒性,要么甚至它们不显示出遗传毒性,即它们显示出与天然存在的背景相当的遗传毒性水平。
[0020]
遗传毒性可以通过如本文进一步所述的测定(toxys,leiden,the netherlands)来进行测量。测定可应用于纯物质或应用于组合物,所述组合物是在本发明的多氮丙啶化合物的制备中获得的直接产物。阳性诱导遗传毒性是指在不存在或存在代谢系统大鼠s9肝提取物的情况下生物标记物bscl2-gfp和rtkn-gfp的诱导水平等于或高于在10%、25%和50%细胞毒性中的至少一者处的2倍。弱阳性诱导遗传毒性是指在不存在或存在基于大鼠s9肝提取物的代谢系统(aroclor1254诱导的大鼠,moltox,boone,nc,usa)的情况下生物标记物bscl2-gfp和rtkn-gfp的诱导水平为在10%、25%和50%细胞毒性中的至少一者处的高于1.5倍并且低于2倍(但是在10%、25%和50%细胞毒性处低于2倍)。与天然存在的背景相当的遗传毒性是指在不存在和存在基于大鼠s9肝提取物的代谢系统(aroclor1254诱导的大鼠,boone,nc,usa)的情况下生物标记物bscl2-gfp和rtkn-gfp的诱导水平小于或等于在10%、25%和50%细胞毒性处的1.5倍。在不存在和存在基于大鼠s9肝提取物的代谢系统(aroclor1254诱导的大鼠,moltox,boone,nc,usa)情况下,遗传毒性报告基因bscl2-gfp和rtkn-gfp的诱导水平优选地小于或等于在10%、25%和50%细胞毒性处的1.5倍。在不存在和存在基于大鼠s9肝提取物的代谢系统(aroclor1254诱导的大鼠,moltox,boone,nc,usa)情况下显示出的诱导水平小于或等于在10%、25%和50%细胞毒性处的1.5倍的物质不是遗传毒性的。
[0021]
对于本文给定的任何范围的所有上边界和/或下边界,除非另外特别指明,否则边界值包括在所述给定的范围内。因此,当描述从x至y时,是指包括x和y以及还有所有中间值。
[0022]
在本说明书中,术语“涂料组合物”涵盖漆、涂料、清漆、粘合剂和油墨组合物,但不限于该列表。术语“脂肪族烃基”是指可选地支链化的烷基、烯基和炔基。术语“脂环族烃基”是指可选地被至少一个脂肪族烃基取代的环烷基和环烯基。术语“芳族烃基”是指可选地被至少一个脂肪族烃基取代的苯环。这些可选取代的脂肪族烃基优选为烷基。具有7个碳原子
的脂环族烃基的示例是环庚基和甲基取代的环己基。具有7个碳原子的芳族烃基的示例是甲基取代的苯基。具有8个碳原子的芳族烃基的示例是二甲苯基和乙基取代的苯基。
[0023]
尽管存在于根据本发明的多氮丙啶化合物中的结构单元(a)可独立地具有不同的r2、r3、r4、r'、r
″
和/或m,但是存在于多氮丙啶化合物中的结构单元(a)优选彼此相同。
[0024]
通常以组合物获得根据本发明的多氮丙啶化合物,在所述组合物中除了多氮丙啶化合物以外,还可存在剩余的起始材料、副产物和/或用于制备多氮丙啶化合物的溶剂。该组合物可包含仅一种根据本发明的多氮丙啶化合物,但是也可以含有多于一种根据本发明的多氮丙啶化合物。例如,当使用多异氰酸酯的混合物作为起始材料时,获得了多氮丙啶化合物的混合物。
[0025]
本发明的氨基甲酸酯氮丙啶化合物含有2至6个结构单元(a),优选2至4个结构单元(a),更优选2或3个结构单元(a)。
[0026]
r2和r4独立地选自h;含有1至8个碳原子、在链中可选地含有一个或多个杂原子(优选选自n、s和o)的直链基团;含有3至8个碳原子的碳原子且在链中可选地含有一个或多个杂原子(优选选自n、s和o)的支链或环状基团;苯基;苄基;或吡啶基。在r2与h不同的情况下,则r2和r3可以是含有3至8个碳原子的同一环状基团的一部分,优选含有3至8个碳原子的同一饱和脂环族烃基的一部分。优选地,r2和r4独立地选自h、含有1至8个碳原子的脂肪族烃基或含有3至8个碳原子的脂环族烃基。更优选地,r2和r4独立地选自h或含有1至4个碳原子的脂肪族烃基。更优选地,r2和r4独立地选自h或含有1至2个碳原子的脂肪族烃基。
[0027]
r3为含有1至8个碳原子并且在链中可选地含有一个或多个杂原子(优选选自n、s和o)的直链基团、含有3至8个碳原子并且在链中可选地含有一个或多个杂原子(优选选自n、s和o)的支链或环状基团、苯基、苄基或吡啶基。r3优选为含有1至8个碳原子的脂肪族烃基、含有3至8个碳原子的脂环族烃基、苯基、苄基或吡啶基。r3更优选为含有1至4个碳原子的脂肪族烃基。
[0028]
在本发明的一个优选实施方式中,r2为h,r3为c2h5并且r4为h。在本发明的另一个更优选的实施方式中,r2为h,r3为ch3并且r4为h或ch3。在本发明的另一个甚至更优选的实施方式中,r2为h,r3为ch3并且r4为h。
[0029]
m为1至6的整数,优选m为1至4,更优选m为1或2,并且最优选m为1。
[0030]
r'为h或含有1至12个碳原子的脂肪族烃基,优选含有1至12个碳原子的烷基。r'优选为h或含有1至4个碳原子的烷基。更优选地,r'为h或含有1至2个碳原子的烷基。最优选地,r'为h。
[0031]
r
″
优选为h、含有1至8个碳原子(更优选1至4个碳原子)的脂肪族烃基、含有5至12个碳原子的脂环族烃基、含有6至12个碳原子的芳族烃基、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(ocr
″″′
hcr
″″′
h)
n-or
″″″
,其中r
″′
为含有1至12个碳原子的脂肪族烃基并且r
″″
为含有1至12个碳原子的脂肪族烃基或含有6至12个碳原子的芳族烃基,n为1至35,优选6至20,r
″″′
独立地为h或甲基并且r
″″″
为含有1至4个碳原子的脂肪族烃基,优选具有1至4个碳原子的烷基,或者r'和r
″
可为含有5至8个碳原子的同一饱和脂环族烃基的一部分。更优选地,r
″
=h、含有1至4个碳原子的脂肪族烃基、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(ocr
″″′
hcr
″″′
h)
n-[0032]
or
″″″
,其中r
″′
为含有1至12个碳原子的烷基,并且r
″″
为含有1至12个碳原子的烷
基,n为1至35,r
″″′
独立地为h或甲基,并且r
″″″
为含有1至4个碳原子的烷基;
[0033]
或者r'和r
″
可为含有5至8个碳原子的同一饱和脂环族烃基的一部分。
[0034]
更优选地,r'为h并且r
″
=含有1至4个碳原子的烷基、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(och2ch2)
n-och3,其中r
″′
优选为含有3至12个碳原子的烷基,更优选具有3至12个碳原子的支链烷基,例如新戊基或新癸基。最优选地,r
″′
为支链的c9烷基。r
″″
优选为含有1至12个碳原子的烷基。r
″″
的非限制性示例为乙基、丁基和2-乙基己基。
[0035]
根据本发明的多氮丙啶化合物的分子量为600道尔顿至5000道尔顿。根据本发明的多氮丙啶化合物的分子量为优选至多3800道尔顿,更优选至多3600道尔顿,更优选至多3000道尔顿,更优选至多1600道尔顿,甚至更优选至多1200道尔顿。根据本发明的多氮丙啶化合物的分子量优选为至少700道尔顿,更优选至少800道尔顿,甚至更优选至少840道尔顿,并且最优选至少1000道尔顿。如本文所用,多氮丙啶化合物的分子量是计算的分子量。所述计算的分子量是通过将多氮丙啶化合物的结构式中存在的所有原子的原子质量相加而获得的。如果多氮丙啶化合物存在于包含多于一种根据本发明的多氮丙啶化合物的组合物中,例如当用于制备多氮丙啶化合物的起始材料中的一种或多种起始材料为混合物时,则可以针对所述组合物中单独存在的每种化合物进行分子量计算。如下文的实验部分中所述,可以使用maldi-tof质谱法测量根据本发明的多氮丙啶化合物的分子量。
[0036]
根据本发明的多氮丙啶化合物包含一个或多个连接链,其中这些连接链中的每一个连接链连接结构单元a中的两个结构单元a。存在于多氮丙啶化合物中的连接链优选由2至300个原子,更优选5至250个原子,最优选6至100个原子组成。连接链的原子优选为c、n、o、s和/或p,优选为c、n和/或o。
[0037]
连接链被定义为连接两个结构单元a的连续原子的最短链。下图示出了根据本发明的多氮丙啶化合物的一个示例,在两个结构单元a之间的连接链。
[0038][0039]
本发明的多氮丙啶化合物中存在的结构单元a中的任何两个结构单元a通过如本
文所定义的连接链连接。因此,本发明的多氮丙啶化合物中存在的每个结构单元a通过如本文所定义的连接链与每个其他结构单元a连接。在根据本发明的多氮丙啶化合物具有两个结构单元a的情况下,多氮丙啶化合物具有一个连接这两个结构单元的此类连接链。在根据本发明的多氮丙啶化合物具有三个结构单元a的情况下,该多氮丙啶化合物具有三个连接链,其中该三个连接链中的每一个连接链将结构单元a与另一个结构单元a连接在一起,即第一结构单元a通过连接链与第二结构单元a连接,并且第一和第二结构单元a均通过它们相应的连接链独立地与第三结构单元a连接。
[0040]
以下附图示出了具有三个结构单元a、三个连接链的多氮丙啶化合物的示例,其中该三个连接链中的每个连接链连接两个结构单元a。
[0041][0042]
具有多于两个结构单元a的根据本发明的多氮丙啶化合物具有根据以下方程式的连接链数量:lc={(an-1)
×
an)}/2,其中lc=多氮丙啶化合物中的连接链的数量,并且an
=多氮丙啶化合物中的结构单元a的数量。因此,例如如果在多氮丙啶化合物中有5个结构单元a,an=5;则这意味着有{(5-1)
×
5}/2=10个连接链。
[0043]
优选地,在结构单元a中的氨基甲酸酯基团的n原子与另一个n原子(其存在于连接链中,或为另一结构单元a的氨基甲酸酯基团的n原子)的之间的连续c原子和可选的o原子的数量为至多9,如在例如以下根据本发明的多氮丙啶化合物中所示。
[0044][0045]
根据本发明的多氮丙啶化合物优选包含一个或多个连接基团(connecting group),其中这些连接基团中的每个连接基团连接结构单元a中的两个结构单元a,其中连接基团被定义为连接两个结构单元a的连续官能团(如所文所定义的官能团)的阵列。在本发明中,连接基团优选由选自由以下项组成的组的至少一个官能团组成:脂肪族烃官能团(优选含有1至8个碳原子)、脂环族烃官能团(优选含有4至10个碳原子)、芳族烃官能团(优选含有6至12个碳原子)、异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、醚官能团、酯官能团、酰胺官能团、碳酸酯官能团、氨基甲酸酯官能团、脲官能团、缩二脲官能团、脲基甲酸酯官能团、脲二酮官能团以及它们的任意组合。
[0046]
下图以粗体示出了根据本发明的多氮丙啶化合物的一个示例的连接基团。在该示例中,连接结构单元a中的两个结构单元a的连接基团由以下连续官能团的阵列组成:脂肪族烃官能团1(直链c6h
12
)、异氰脲酸酯2(环状c3n3o3)官能团和脂肪族烃官能团3(直链c6h
12
)。
[0047][0048]
下图以粗体示出了根据本发明的多氮丙啶化合物的以下示例的连接基团。在该示例中,连接两个结构单元a的连接基团由以下连续官能团的阵列组成:脂肪族烃官能团1(直链c6h
12
)、异氰脲酸酯2(环状c3n3o3)和脂肪族烃官能团3(直链c6h
12
)。
[0049][0050]
本发明的多氮丙啶化合物中存在的结构单元a中的任何两个结构单元a通过如本文所定义的连接基团连接。因此,本发明的多氮丙啶化合物中存在的每个结构单元a使用如在本发明中所定义的连接基团与每个其他结构单元a连接。在根据本发明的多氮丙啶化合物具有两个结构单元a的情况下,多氮丙啶化合物具有一个连接这两个结构单元的此类连接基团。在根据本发明的多氮丙啶化合物具有三个结构单元a的情况下,该多氮丙啶化合物具有三个此类连接基团,其中该三个连接基团中的每一个连接基团将结构单元a与另一个结构单元a连接在一起。
[0051]
下图示出了对于具有三个结构单元a的多氮丙啶化合物的示例来说的三个连接基团,其中该三个连接基团中的每个连接基团连接两个结构单元a。一个连接基团由以下连续官能团的阵列组成:连接被标记为a1和a2的结构单元a的脂肪族烃官能团1(直链c6h
12
)、异氰脲酸酯2(环状c3n3o3)和脂肪族烃官能团3(直链c6h
12
)。对于被标记为a1和a3的结构单元a
之间的连接,连接基团由以下连续官能团的阵列组成:脂肪族烃官能团1(直链c6h
12
)、异氰脲酸酯2(环状c3n3o3)和脂肪族烃官能团4(直链c6h
12
),而对于被标记为a2和a3的结构单元a之间的连接,连接基团由以下连续官能团的阵列组成:脂肪族烃官能团3(直链c6h
12
)、异氰脲酸酯2(环状c3n3o3)和脂肪族烃官能团4(直链c6h
12
)。
[0052][0053][0054]
下图示出了根据本发明的多氮丙啶化合物的另一示例,其中两个结构单元a之间有连接链。
[0055][0056]
在该示例中,连接两个结构单元a的连接基团由以下连续官能团的阵列组成:脂肪族烃官能团1(支链c3h6)、芳族烃官能团2(苯环)和脂肪族烃官能团3(支链c3h6)。
[0057][0058]
在根据本发明的多氮丙啶化合物的另一个示例中,连接两个结构单元a的连接基团由以下连续官能团的阵列组成:脂肪族烃官能团1(直链c6h
12
)、脲二酮2(环状c2n2o2)和脂肪族烃官能团3(直链c6h
12
)。
[0059][0060]
优选地,连接基团由选自由以下项组成的组的至少一个官能团组成:脂肪族烃官
能团(优选含有1至8个碳原子)、脂环族烃官能团(优选含有4至10个碳原子)、芳族烃官能团(优选含有6至12个碳原子)、异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、氨基甲酸酯官能团、脲官能团、缩二脲官能团以及它们的任何组合。连接基团优选含有异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、缩二脲官能团、脲基甲酸酯官能团或脲二酮官能团。更优选地,连接基团含有异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团。为了清楚起见,多氮丙啶化合物可以从一种或多种合适的化合物b与混杂的异氰脲酸酯(例如hdi/ipdi异氰脲酸酯)的反应产物获得,从而产生具有由以下连续官能团的阵列组成的连接基团的多氮丙啶化合物:直链c6h
12
(即具有6个碳原子的脂肪族烃官能团)、异氰脲酸酯官能团(环状c3n3o3)和
[0061][0062]
(即具有9个碳原子的脂环族烃官能团和具有1个碳原子的脂肪族烃官能团)。
[0063]
术语“脂肪族烃官能团”是指可选地支链化的烷基、烯基和炔基。尽管c原子的可选支链为连接基团的组成部分,但是它们不是连接链的组成部分。术语“脂环族烃基官能团”是指可选地被至少一个脂肪族烃基取代的环烷基和环烯基。尽管可选的脂肪族烃基取代基是连接基团的组成部分,但是它们不是连接链的组成部分。术语“芳族烃官能团”是指可选地被至少一个脂肪族烃基取代的苯环。可选地被取代的脂肪族烃基优选为烷基。尽管可选的脂肪族烃基取代基是连接基团的组成部分,但是它们不是连接链的组成部分。
[0064]
在连接基团上,一个或多个取代基可以作为连接基团上的侧基存在,如在例如以下多氮丙啶化合物中以粗体所示。这些侧基不是连接基团的组成部分。
[0065][0066]
氮丙啶基具有以下结构式:
[0067][0068]
异氰脲酸酯官能团被定义为
[0069]
亚氨基噁二嗪二酮官能团被定义为
[0070]
脲基甲酸酯官能团被定义为
[0071]
脲二酮官能团被定义为
[0072]
缩二脲官能团被定义为
[0073]
在本发明的一个优选实施方式中,本发明的多氮丙啶化合物中存在的连接基团由以下官能团组成:至少一个脂肪族烃官能团和/或至少一个脂环族烃官能团和可选的至少一个芳族烃官能团和可选的异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团或脲基甲酸酯官能团或脲二酮官能团。优选地,本发明的多氮丙啶化合物中存在的连接基团由以下官能团组成:至少一个脂肪族烃官能团和/或至少一个脂环族烃官能团和可选的至少一个芳族烃官能团和可选的异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团。获得此类多氮丙啶化合物的非常合适的方式是使具有以下结构式的化合物b与具有脂肪族反应性的多异氰酸酯反应:
[0074][0075]
术语“具有脂肪族反应性的多异氰酸酯”是指这样的化合物,在所述化合物中所有异氰酸酯基团直接键合至脂肪族或脂环族烃基团,而与是否还存在芳族烃基团无关。具有脂肪族反应性的多异氰酸酯可为具有脂肪族反应性的多异氰酸酯的混合物。当与类似但是基于具有芳族反应性的聚异氰酸酯的化合物相比时,基于具有脂肪族反应性的聚异氰酸酯的化合物随时间推移黄化的趋势降低。术语“具有芳族反应性的多异氰酸酯”是指这样的化合物,在所述化合物中所有异氰酸酯基团都直接键合至芳族烃基团,而与是否还存在脂肪族或脂环族基团无关。优选的具有脂肪族反应性的多异氰酸酯为1,5-五亚甲基二异氰酸酯pdi、1,6-六亚甲基二异氰酸酯hdi、异佛尔酮二异氰酸酯ipdi、4,4'-二环己基甲烷二异氰酸酯h12mdi、2,2,4-三甲基六亚甲基二异氰酸酯、2,4,4-三甲基六亚甲基二异氰酸酯、四甲基二甲苯二异氰酸酯tmxdi(所有异构体)和更高分子量的变体,如例如它们的异氰脲酸酯或亚氨基噁二嗪二酮。在该实施方式中,优选地,连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、芳族烃官能团和脂肪族烃官能团(例如,当使用tmxdi制备多氮丙啶化合物时),或者连接基团由以下连续官能团的阵列组成:脂环族烃官能团、脂肪族烃官能团和
脂环族烃官能团(例如,当使用h12mdi制备多氮丙啶化合物时),或者更优选地,连接基团由以下连续官能团的阵列组成:脂肪族烃官能团,异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团,和脂肪族烃官能团。最优选地,在该实施方式中,连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、异氰脲酸酯官能团和脂肪族烃官能团(例如,当使用1,6-六亚甲基二异氰酸酯的异氰脲酸酯和/或1,5-五亚甲基二异氰酸酯的异氰脲酸酯制备多氮丙啶化合物时)。
[0076]
在本发明的另一个实施方式中,根据本发明的多氮丙啶化合物为根据以下结构式:
[0077][0078]
其中z为通过去除分子的异氰酸酯反应性基团xh获得的分子残基;
[0079]
q为2至6的整数;
[0080]
i为不同基团d的索引,并且为1至q的整数;
[0081]
d
i
独立地具有以下结构式
[0082][0083]
其中x为nr
11
、s或o,其中r
11
为h或具有1至4个碳原子的烷基;
[0084]
y为芳族烃基、脂肪族烃基、脂环族烃基或它们的组合;
[0085]
j为1至p的整数;
[0086]
p为0至10的整数。
[0087]
m、r'、r
″
、r1、r2、r3和r4如上所定义。在本发明的该实施方式中,多氮丙啶化合物含有2至6个d
i
基团。虽然结构单元d
i
可以独立地为相同或不同的,但是结构单元d
i
优选彼此相同。
[0088]
异氰酸酯反应性基团xh在本文中被定义为羟基(x为o)、伯胺(x为nh)或仲胺(x为nr
11
,其中r
11
为具有1至4个碳原子的烷基)或硫醇(x为s)。优选的异氰酸酯反应性基团xh为羟基(x为o)、伯胺(x为nh)或仲胺(x为nr
11
,其中r
11
为具有1至4个碳原子的烷基)。更优选的异氰酸酯反应性基团xh为羟基(x为o)和伯胺(x为nh)。从中去除异氰酸酯反应性基团以获得z的所述分子优选为二醇、三醇、具有末端异氰酸酯反应性基团的聚醚、具有末端异氰酸酯反应性基团的聚酰胺、具有末端异氰酸酯反应性基团的聚碳酸酯、或具有末端异氰酸酯反应性基团的聚硅氧烷,具有末端异氰酸酯反应性基团的聚硅氧烷中的末端异氰酸酯反应性基团通过至少一个碳原子与所述硅氧烷连接。在z为通过去除二醇或三醇的异氰酸酯反应性基团xh而获得的分子残基的情况下,该异氰酸酯反应性基团xh为羟基并且因此x为o。在z为通过去除具有末端异氰酸酯反应性基团的聚醚或具有末端异氰酸酯反应性基团的聚酰胺的异氰酸酯反应性基团xh而获得的分子残基的情况下,该异氰酸酯反应性基团xh优选为nh2(因此x为nh)或oh(因此x为o),并且更优选地异氰酸酯反应性基团xh为oh(因此x为o)。在z为通过去除具有末端异氰酸酯反应性基团的聚碳酸酯的异氰酸酯反应性基团xh而获得的分子残基的情况下,异氰酸酯反应性基团优选为oh并且因此x为o。
[0089]
在j大于1的情况下,则z可为相同或不同的。
[0090]
优选地,q为2或3,并且更优选地,q为1。
[0091]
优选地,p为0至10,更优选0至5,最优选0至3的整数。
[0092]
在该实施方式中,对于所有d
i
,p最优选为0,并且因此d
i
独立地具有以下结构式,
[0093][0094]
其中m、r'、r
″
、r1、r2、r3和r4如上所定义。优选地m为1。
[0095]
虽然结构单元d
i
可以独立地为相同或不同的,但是结构单元d
i
优选彼此相同。
[0096]
在多氮丙啶化合物中存在的环状结构(除氮丙啶以外)的总量优选为至多3,因为这导致与当存在较高量的环状结构时相比较低的粘度。较低的粘度更易于处理和/或需要较少的助溶剂来使化合物更易于处理。如果多氮丙啶化合物在环境温度下为固体,则当溶解此类多氮丙啶时,具有多于三个环状结构的多氮丙啶化合物可导致更多的困难。在多氮丙啶化合物中存在的环状结构(除氮丙啶以外)的总量更优选为0至2,甚至更优选为1或2,并且最优选为1,其优选为异氰脲酸酯或亚氨基噁二嗪二酮。
[0097]
根据本发明的多氮丙啶化合物优选含有至少5重量%,更优选至少5.5重量%,更优选至少6重量%,更优选至少9重量%,更优选至少12重量%并且优选小于25重量%,优选小于20重量%的氨基甲酸酯键。根据本发明的多氮丙啶化合物的氮丙啶当量重量(多氮丙啶化合物的分子量除以多氮丙啶化合物中存在的氮丙啶基团的数量)优选为至少200道尔顿,更优选至少230道尔顿并且甚至更优选至少260道尔顿,并且优选至多2500道尔顿,更优选至多1000道尔顿并且甚至更优选至多500道尔顿。
[0098]
如果需要的话,可以用0.1重量%至1重量%的叔胺,优选β-羟基胺(如amietol m21或amietol m-12)来稳定化多氮丙啶化合物。
[0099]
根据本发明的多氮丙啶化合物优选通过使至少多异氰酸酯与具有以下结构式的化合物b反应而获得:
[0100][0101]
其中化合物b与多异氰酸酯的摩尔比为2至6,更优选2至4,并且最优选2至3,并且其中m、r'、r
″
、r1、r2、r3和r4如上面所定义。使多异氰酸酯与化合物b反应可通过以下方式进行:在例如锡催化剂(例如月桂酸二丁锡)或铋催化剂(例如新癸酸铋)的存在下,在从0℃至110℃、更合适从20℃至110℃、更合适从40℃至95℃、甚至更合适从60℃至85℃的范围内的温度下使当量的多异氰酸酯与化合物b接触。可以使用溶剂,例如二甲基甲酰胺dmf、丙酮和/或甲基乙基酮。该多异氰酸酯含有至少2个异氰酸酯基,优选平均至少2.5个异氰酸酯基,更优选平均至少2.8个异氰酸酯基。多异氰酸酯的混合物也可以用作起始材料。优选的多异氰酸酯是具有脂肪族反应性的多异氰酸酯。术语“具有脂肪族反应性的多异氰酸酯”是意指这样的化合物,在所述化合物中所有异氰酸酯基团直接键合至脂肪族或脂环族烃基
团,而与是否还存在芳族烃基团无关。具有脂肪族反应性的多异氰酸酯可为具有脂肪族反应性的多异氰酸酯的混合物。优选的具有脂肪族反应性的多异氰酸酯为1,5-五亚甲基二异氰酸酯pdi、1,6-六亚甲基二异氰酸酯hdi、异佛尔酮二异氰酸酯ipdi、4,4'-二环己基甲烷二异氰酸酯h12mdi、2,2,4-三甲基六亚甲基二异氰酸酯、2,4,4-三甲基六亚甲基二异氰酸酯、对四甲基二甲苯二异氰酸酯(p-tmxdi)及其间位异构体,以及更高分子量的变体,例如它们的异氰脲酸酯或亚氨基噁二嗪二酮或脲基甲酸酯或脲二酮。更优选的具有脂肪族反应性的多异氰酸酯为4,4'-二环己基甲烷二异氰酸酯h12mdi、m-tmxdi、1,6-六亚甲基二异氰酸酯的异氰脲酸酯或亚氨基噁二嗪二酮或脲基甲酸酯或脲二酮和1,5-五亚甲基二异氰酸酯的异氰脲酸酯。合适的含hdi的亚氨基噁二嗪二酮三聚体为可从covestro获得的n3900。合适的含有hdi的脲基甲酸酯为可从covestro获得的xp2860。合适的含有hdi的脲二酮为可从covestro获得的n3400。合适的基于hdi异氰脲酸酯三聚体例如可以从covestro(n3600)、vencorex(tolonate
tm hdt lv)、asahi kasei(duranate
tm tpa-100)、evonik(ht 2500/lv)和tosoh(hxr lv)获得。制备化合物(b)和衍生物的方法是本领域中已知的。例如,s.lesniak、m.rachwalski、s.jarzynski、e.obijalska tetrahedron asymm.2013,24 1336-1340描述了1-(2-甲基吖丙啶-1-基)丙-2-醇的合成。a.baklien,m.v.leeding,j.kolm aust.j.chem.1968,21,1557-1570描述了1-(吖丙啶-1-基)丙-2-醇的合成。用于制备化合物b的优选氮丙啶化合物为丙烯亚胺和乙基氮丙啶。乙基氮丙啶的合成例如描述于ep0227461b1中。用于制备化合物b的最优选的氮丙啶化合物为丙烯亚胺。
[0102]
优选通过使至少非oh官能的单环氧化物化合物与具有以下结构式(c)的氮丙啶化合物反应来获得化合物b:
[0103][0104]
其中r1、r2、r3和r4如上所定义。非oh官能的单环氧化物可为不同的非oh官能的单环氧化物的混合物。非oh官能的单环氧化物的非限制性示例为环氧乙烷、环氧丙烷、2-乙基环氧乙烷、正丁基缩水甘油醚、2-乙基己基缩水甘油醚、苯基缩水甘油醚、4-叔丁基苯基2,3-环氧丙基醚(=叔丁基苯基缩水甘油醚)、甲酚缩水甘油醚(邻位或对位)以及新癸酸缩水甘油酯。非oh官能的单环氧化物优选选自由以下项组成的组:环氧乙烷(cas编号75-21-8)、环氧丙烷(cas编号75-56-9)、2-乙基环氧乙烷(cas编号106-88-7)、正丁基缩水甘油醚(cas编号2426-08-6)、2-乙基己基缩水甘油醚(cas编号2461-15-6)、新癸酸缩水甘油酯(cas编号26761-45-5)以及它们的任何混合物。更优选地,非oh官能的单环氧化物选自由以下项组成的组:环氧丙烷(cas编号75-56-9)、2-乙基环氧乙烷(cas编号106-88-7)、正丁基缩水甘油醚(cas编号2426-08-6)、2-乙基己基缩水甘油醚(cas编号2461-15-6)、新癸酸缩水甘油酯(cas编号26761-45-5)以及它们的任何混合物。
[0105]
根据本发明的多氮丙啶化合物优选以包括至少以下步骤(i)和(ii)的方法获得:
[0106]
(i)使具有式(c)的氮丙啶与至少非oh官能的单环氧化物化合物反应以获得化合
物b,以及
[0107]
(ii)使化合物b与多异氰酸酯反应。
[0108]
步骤(i)可以例如通过在大气压下在从20℃至110℃、更合适从40℃至95℃、甚至更适合从60℃至85℃的范围中的温度下使一当量的环氧化物化合物与一当量的氮丙啶接触来进行。在步骤(i)中获得的加合物(化合物(b))与多异氰酸酯的反应(步骤(ii))可以例如通过以下方式进行:在例如锡催化剂(例如月桂酸二丁锡)的存在下,在大气压下在从20℃至110℃、更合适从40℃至95℃范围内的温度下使当量的多异氰酸酯与所述加合物接触。
[0109]
在一个优选的实施方式中,根据本发明的多氮丙啶化合物优选含有相对于多氮丙啶化合物的优选至少0.1重量%,更优选至少6重量%,更优选至少10重量%的量并且优选小于45重量%,更优选小于25重量%,最优选小于16重量%的量的聚氧乙烯(-o-ch2-ch2-)
x
基团、聚氧丙烯(-o-chch3-ch2-)
x
基团和/或聚四氢呋喃(-o-ch2-ch2-ch2-ch2)
x
基团。优选地,多氮丙啶化合物含有相对于多氮丙啶化合物的优选至少0.1重量%,更优选至少6重量%,更优选至少10重量%的量并且优选小于45重量%,更优选小于25重量%并且最优选小于16重量%的量的聚氧乙烯(-o-ch2-ch2-)
x
基团。为了清楚起见,应当注意的是,在r'=h并且r
″
=h的情况下或者在r'=h并且r
″
=ch3的情况下,一个氧乙烯基或相应地一个氧丙烯基存在于结构单元(a)中并且随后以如本文所定义的优选量的氧乙烯基或氧丙烯基被包含。含有聚氧乙烯(-o-ch2-ch2-)
x
基团的多氮丙啶化合物优选为至少化合物(b)、多异氰酸酯和烷氧基聚(乙二醇)(优选甲氧基聚(乙二醇)(mpeg))和/或聚(乙二醇)的反应产物。反应产物可以通过使至少化合物(b)、多异氰酸酯和烷氧基聚(乙二醇)和/或聚(乙二醇)反应而获得。反应产物还可以通过使多异氰酸酯与烷氧基聚(乙二醇)和/或聚(乙二醇)反应并使如此获得的化合物与化合物(b)反应而获得。反应产物还可以通过使化合物(b)与多异氰酸酯反应并使如此获得的化合物与烷氧基聚(乙二醇)和/或聚(乙二醇)反应而获得。
[0110]
在如上面所定义的多氮丙啶化合物中数均分子量m
n
高于2200道尔顿、优选m
n
高于1600道尔顿的烷氧基聚(乙二醇)(优选甲氧基聚(乙二醇)(mpeg))和/或聚(乙二醇)(peg)链的量优选小于35重量%,更优选小于15重量%,更优选小于5重量%,并且最优选为0重量%。存在于多氮丙啶化合物中的甲氧基聚(乙二醇)(mpeg)和/或聚(乙二醇)(peg)链的m
n
优选低于1100道尔顿,更优选低于770道尔顿,并且最优选低于570道尔顿。
[0111]
根据本发明的优选的多氮丙啶化合物的示例为
[0112][0113][0114]
本发明的另一方面是一种交联剂组合物,所述交联剂组合物包含如上面所定义的至少一种多氮丙啶化合物,并且还包含至少一种附加组分,例如用于制备根据本发明的多氮丙啶化合物的溶剂、剩余的起始材料和/或副产物。该交联剂组合物可仅包含一种根据本
发明的多氮丙啶化合物,但是也可以含有多于一种根据本发明的多氮丙啶化合物。例如,当使用多异氰酸酯的混合物作为制备多氮丙啶的起始材料时,获得了多氮丙啶化合物的混合物。在已经获得根据本发明的多氮丙啶化合物之后,可以分离根据本发明的多氮丙啶化合物,反应产物可以不经进一步纯化就使用,或者可以从在本发明的多氮丙啶化合物的制备中获得的组合物中去除用于制备所述多氮丙啶化合物的溶剂。相对于组合物的总量,在交联剂组合物中根据本发明的多氮丙啶化合物的量通常为至少10重量%,往往通常至少15重量%并且最通常至少25重量%。相对于交联剂组合物的总量,在交联剂组合物中根据本发明的多氮丙啶化合物的量为优选至少60重量%,更优选至少80重量%,并且最优选至少99重量%。交联剂组合物中多氮丙啶化合物的分子量在600道尔顿至5000道尔顿的范围中。优选的分子量如上所述,并且多氮丙啶化合物的分子量是如下文的实验部分中所述使用maldi-tof-ms测定的。maldi-tof-ms是指基质辅助的激光解吸电离飞行时间质谱。
[0115]
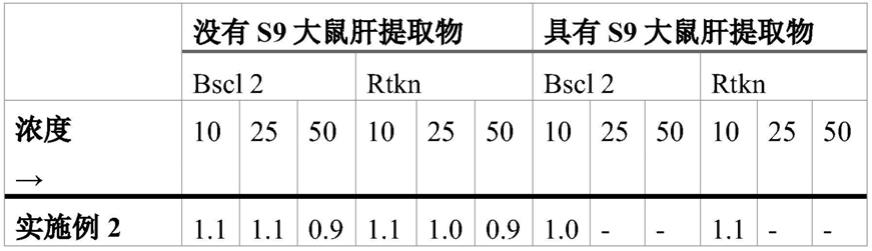
相对于交联剂组合物的总重量,根据本发明的交联剂组合物中存在的分子量低于250道尔顿,更优选低于350道尔顿,甚至更优选低于450道尔顿,甚至更优选低于550道尔顿并且甚至更优选低于580道尔顿的氮丙啶官能分子的量优选低于5重量%,更优选低于2重量%,更优选低于1重量%,更优选低于0.5重量%并且最优选低于0.1重量%,其中所述分子量是如以下实验部分中所述使用lc-ms测定的。
[0116]
组合物中每个含氮丙啶基的分子的氮丙啶基的平均数量为优选至少1.8个,更优选至少2个,更优选至少2.2个并且优选小于10个,更优选小于6个并且最优选小于4个。最优选地,组合物中每个含氮丙啶基的分子中的氮丙啶基的平均数量为2.2至3。相对于交联剂组合物中存在的根据本发明的多氮丙啶化合物的总重量,氨基甲酸酯键的计算平均量为至少5重量%,更优选至少5.5重量%,更优选至少6重量%,更优选至少9重量%,更优选至少12重量%,并且优选小于25重量%,优选小于20重量%。
[0117]
鉴于根据本发明的多氮丙啶化合物的潜在水敏感性,交联剂组合物优选不含显著量的水,并且更优选不含水。不含显著量的水是指小于15重量%,优选小于5重量%,更优选小于1重量%并且最优选小于0.1重量%。鉴于根据本发明的多氮丙啶化合物的潜在水敏感性,水优选不是故意添加到组合物中的(即,在用于制备根据本发明的多氮丙啶化合物的化合物中可以存在少量的水)。
[0118]
根据本发明的多氮丙啶化合物优选在25℃下的布氏粘度为至少500mpa.s,更优选至少1200mpa.s,更优选至少3000mpa.s,并且在25℃下的布氏粘度为优选至多1000000mpa.s,更优选至多100000mpa.s,更优选至多30000mpa.s,更优选为10000mpa.s并且最优选5000mpa.s。如本文所用,布氏粘度是根据iso 2555-89测定的。在一个替代实施方式中,用具有锭子s63的brookfield在25℃下在80%固体、20%二甲基甲酰胺(dmf)中测量多氮丙啶的粘度。如根据该方法测量的粘度优选在300mpa.s至20000mpa.s的范围内,更优选在500mpa.s至12000mpa.s的范围内,并且最优选在700mpa.s至3000mpa.s的范围内。
[0119]
根据本发明的多氮丙啶化合物或包含至少一种如上面所定义的多氮丙啶化合物的交联剂组合物可以有利地用作交联剂,以使羧酸官能聚合物交联、优选地溶解和/或分散在水性介质中的羧酸官能聚合物交联。
[0120]
本发明的另一方面是一种双组分体系,所述双组分体系包含彼此分开并且不同的第一组分和第二组分,其中所述第一组分包含溶解和/或分散在水性介质中的羧酸官能聚
or
″″″
,其中r
″′
为含有1至12个碳原子的脂肪族烃基并且r
″″
为含有1至12个碳原子的脂肪族烃基或含有6至12个碳原子的芳族烃基,n为1至35,r
″″′
独立地为h或含有1至12个碳原子的脂肪族烃基,并且r
″″″
为含有1至4个碳原子的脂肪族烃基;
[0132]
其中r'和r
″
可为含有5至8个碳原子的同一饱和脂环族烃基的一部分;
[0133]
m为1至6的整数;
[0134]
多氮丙啶化合物还包含一个或多个连接链,其中这些连接链中的每一个连接链连接所述多氮丙啶化合物中存在的结构单元a中的两个结构单元a,其中所述连接链优选由2至300个原子组成,更优选由5至250个原子组成,最优选由6至100个原子组成;并且
[0135]
其中多氮丙啶化合物的分子量在600道尔顿至5000道尔顿的范围内,其中多氮丙啶化合物的分子量是计算分子量或如下面的实验部分中所述使用maldi-tof质谱法测量的。
[0136]
[2]根据实施方式[1]所述的多氮丙啶化合物,其中所述多氮丙啶化合物含有2至4个结构单元(a),优选2或3个结构单元(a),其中所述多氮丙啶化合物中存在的结构单元(a)优选彼此相同。
[0137]
[3]根据实施方式[1]或[2]所述的多氮丙啶化合物,其中r2和r4独立地选自h或含有1至8个碳原子的脂肪族烃基,优选地r2和r4独立地选自h或含有1至4个碳原子的脂肪族烃基,优选地r2和r4独立地选自h或含有1至2个碳原子的脂肪族烃基。
[0138]
[4]根据[1]至[3]中的任何实施方式所述的多氮丙啶化合物,其中r3为含有1至8个碳原子的脂肪族烃基,优选r3为含有1至4个碳原子的脂肪族烃基。
[0139]
[5]根据[1]至[4]中的任何实施方式所述的多氮丙啶化合物,其中r2为h,r3为c2h5并且r4为h。
[0140]
[6]根据[1]至[4]中的任何实施方式所述的多氮丙啶化合物,其中r2为h,r3为ch3并且r4为h。
[0141]
[7]根据[1]至[4]中的任何实施方式所述的多氮丙啶化合物,其中r2为h,r3为ch3并且r4为ch3。
[0142]
[8]根据[1]至[7]中的任何实施方式所述的多氮丙啶化合物,其中m为1。
[0143]
[9]根据[1]至[8]中的任何实施方式所述的多氮丙啶化合物,其中r'为h或含有1至12个碳原子的烷基,优选地r'为h或含有1至4个碳原子的烷基,更优选地r'为h或含有1至2个碳原子的烷基。
[0144]
[10]根据[1]至[9]中的任何实施方式所述的多氮丙啶化合物,其中r
″
为h、含有1至8个碳原子(更优选1至4个碳原子)的脂肪族烃基、含有5至12个碳原子的脂环族烃基、含有6至12个碳原子的芳族烃基、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(ocr
″″′
hcr
″″′
h)
n-or
″″″
,其中r
″′
为含有1至12个碳原子的脂肪族烃基并且r
″″
为含有1至12个碳原子的脂肪族烃基或含有6至12个碳原子的芳族烃基,n为1至35、优选6至20,r
″″′
独立地为h或甲基并且r
″″″
为含有1至4个碳原子的脂肪族烃基,优选具有1至4个碳原子的烷基,或者r'和r
″
可为含有5至8个碳原子的同一饱和脂环族烃基的一部分。更优选地,r'为h并且r
″
=含有1至4个碳原子的烷基、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(och2ch2)
n-och3,其中r
″′
优选为含有3至12个碳原子的烷基,更优选具有3至12个碳原子的支链烷基,最优选r
″′
为支链的c9烷基;r
″″
优选为含有1至12个碳原子的烷基。
[0145]
[11]根据[1]至[10]中的任何实施方式所述的多氮丙啶化合物,其中所述连接链的原子为c、n、o、s和/或p,优选地c、n和/或o。
[0146]
[12]根据[1]至[11]中的任何实施方式所述的多氮丙啶化合物,其中在结构单元a中的氨基甲酸酯的n原子与另一个n原子(其存在于连接链中或为另一结构单元a的氨基甲酸酯的n原子)的之间的连续c原子和可选的o原子的数量为至多9。
[0147]
[13]根据[1]至[12]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物包含一个或多个连接基团,其中这些连接基团中的每个连接基团连接多氮丙啶化合物中存在的结构单元a中的两个结构单元a,其中所述连接基团由选自以下的至少一个官能团组成:
[0148]
脂肪族烃官能团(优选含有4至10个碳原子)、脂环族烃官能团(优选含有4至10个碳原子)、芳族烃官能团(优选含有6至12个碳原子)、异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、醚官能团、酯官能团、酰胺官能团、碳酸酯官能团、氨基甲酸酯官能团、脲官能团、缩二脲官能团、脲基甲酸酯官能团、脲二酮官能团以及它们的任意组合。
[0149]
[14]根据[1]至[13]中的任何实施方式所述的多氮丙啶化合物,其中所述连接基团由选自由以下项组成的组的至少一个官能团组成:脂肪族烃官能团(优选含有1至8个碳原子)、脂环族烃官能团(优选含有4至10个碳原子)、芳族烃官能团(优选含有6至12个碳原子)、异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、氨基甲酸酯官能团、脲官能团、缩二脲官能团以及它们的任何组合。
[0150]
[15]根据[1]至[14]中的任何实施方式所述的多氮丙啶化合物,其中所述连接基团优选含有异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团或缩二脲官能团。
[0151]
[16]根据[1]至[15]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物的分子量为至少700道尔顿,更优选至少800道尔顿,甚至更优选至少840道尔顿,甚至更优选至少1000道尔顿,并且优选至多3800道尔顿,更优选至多3600道尔顿,更优选至多3000道尔顿,更优选至多1600道尔顿,甚至更优选至多1200道尔顿。
[0152]
[17]根据[1]至[16]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物的连接基团由以下官能团组成:至少一个脂肪族烃官能团和/或至少一个脂环族烃官能团和可选的至少一个芳族烃官能团和可选的异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团。
[0153]
[18]根据[1]至[17]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物是通过使具有以下结构式的化合物b与具有脂肪族反应性的多异氰酸酯反应而获得的:
[0154][0155]
[19]根据实施方式[18]所述的多氮丙啶化合物,其中具有脂肪族反应性的多异氰酸酯选自1,5-五亚甲基二异氰酸酯pdi、1,6-六亚甲基二异氰酸酯hdi、异佛尔酮二异氰酸酯ipdi、4,4'-二环己基甲烷二异氰酸酯h12mdi、2,2,4-三甲基六亚甲基二异氰酸酯、2,4,4-三甲基六亚甲基二异氰酸酯、四甲基二甲苯二异氰酸酯tmxdi(所有异构体)和更高分子
量的变体,如例如它们的异氰脲酸酯或亚氨基噁二嗪二酮。
[0156]
[20]根据[13]至[18]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物的连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、芳族烃官能团和脂肪族烃官能团(例如当使用tmxdi制备多氮丙啶化合物时),或者所述连接基团由以下连续官能团的阵列组成:脂环族烃官能团、脂肪族烃官能团和脂环族烃官能团(例如当使用h12mdi制备多氮吡啶化合物时),或者所述连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团和脂肪族烃官能团,或者所述连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、异氰脲酸酯官能团和脂肪族烃官能团(例如当使用1,6-六亚甲基二异氰酸酯的异氰脲酸酯和/或1,5-五亚甲基二异氰酸酯的异氰脲酸酯制备多氮丙啶化合物时)。
[0157]
[21]根据[1]至[12]中的任何实施方式所述的多氮丙啶化合物,其中根据本发明的多氮丙啶化合物为根据以下结构式:
[0158][0159]
其中z为通过去除分子的异氰酸酯反应性基团xh获得的分子残基;
[0160]
q为2至6的整数;
[0161]
i为不同基团d
i
的索引,并且为1至q的整数;
[0162]
d
i
独立地具有以下结构式
[0163][0164]
其中x为nr
11
、s或o,其中r
11
为h或具有1至4个碳原子的烷基;y为芳族烃基、脂肪族烃基、脂环族烃基或它们的组合;j为1至p的整数;z为通过去除分子的异氰酸酯反应性基团xh获得的分子残基;p为0至10的整数,m、r'、r
″
、r1、r2、r3和r4如上面所定义,其中异氰酸酯反应性基团xh在本文中被定义为羟基(x为o)、伯胺(x为nh)或仲胺(x为nr
11
,其中r
11
为具有1至4个碳原子的烷基)或硫醇(x为s),优选的异氰酸酯反应性基团xh为羟基(x为o)、伯胺(x为nh)或仲胺(x为nr
11
,其中r
11
为具有1至4个碳原子的烷基),更优选的异氰酸酯反应性基团xh为羟基(x为o)和伯胺(x为nh)。优选地,从中去除异氰酸酯反应性基团以获得z的分子优选为二醇、三醇、具有末端异氰酸酯反应性基团的聚醚、具有末端异氰酸酯反应性基团的聚酰胺、具有末端异氰酸酯反应性基团的聚碳酸酯或具有末端异氰酸酯反应性基团的聚硅氧烷,具有末端异氰酸酯反应性基团的聚硅氧烷中的末端异氰酸酯反应性基团通过至少一个碳原子与硅氧烷连接,在z为通过去除二醇或三醇的异氰酸酯反应性基团xh获得的分子残基的情况下,所述异氰酸酯反应性基团xh为羟基并且因此x为o,在z为通过去除具有末端异氰酸酯反应性基团的聚醚的异氰酸酯反应性基团xh或具有末端异氰酸酯反应性基团的聚酰胺的异氰酸酯反应性基团xh而获得的分子残基的情况下,所述异氰酸酯反应性基团xh优选为nh2(因此x为nh)或oh(因此x为o),并且更优选异氰酸酯反应性基团xh为oh(因此x为o),在z为通过去除具有末端异氰酸酯反应性基团的聚碳酸酯的异氰酸酯反应性基团xh而获得的分子残基的情况下,所述异氰酸酯反应性基团优选为oh并且因此x为o,在j大于1的
情况下,则z可为相同或不同的,优选地q为2或3,并且更优选地,q为1,优选地p为0至10,更优选0至5,甚至更优选0至3的整数,并且最优选地,p为0并且m为0。
[0165]
[22]一种多氮丙啶化合物,所述多氮丙啶化合物具有2至6个、优选2至4个、更优选2至3个如在实施方式[1]中所定义的结构单元(a),其中r1、r2、r3、r4、r'、r
″
和m如在实施方式[1]至[10]中任一项所定义,其中多氮丙啶化合物的分子量为600道尔顿至5000道尔顿,优选至少700道尔顿,更优选至少800道尔顿,甚至更优选至少840道尔顿并且优选至多3800道尔顿,更优选至多3600道尔顿,更优选至多3000道尔顿,更优选至多1600道尔顿,甚至更优选至多1200道尔顿,并且其中多氮丙啶化合物还包含一个或多个连接基团,其中这些连接基团中的每个连接基团连接结构单元a中的两个结构单元a,其中所述连接基团由选自由以下项组成的组的至少一个官能团组成:脂肪族烃官能团(优选含有1至8个碳原子)、脂环族烃官能团(优选含有4至10个碳原子)、芳族烃官能团(优选含有6至12个碳原子)、异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、醚官能团、酯官能团、酰胺官能团、碳酸酯官能团、氨基甲酸酯官能团、脲官能团、缩二脲官能团、脲基甲酸酯官能团、脲二酮官能团以及它们的任意组合。
[0166]
[23]根据实施方式[22]所述的多氮丙啶化合物,其中所述多氮丙啶化合物的连接基团由选自由以下项组成的组的至少一个官能团组成:脂肪族烃官能团(优选含有1至8个碳原子)、脂环族烃官能团(优选含有4至10个碳原子)、芳族烃官能团(优选含有6至12个碳原子)、异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团、氨基甲酸酯官能团、脲官能团、缩二脲官能团以及它们的任何组合。
[0167]
[24]根据实施方式[22]或[23]所述的多氮丙啶化合物,其中所述连接基团优选含有异氰脲酸酯官能团、亚氨基噁二嗪二酮官能团或缩二脲官能团。
[0168]
[25]根据[22]至[23]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物的连接基团由以下官能团组成:至少一个脂肪族烃官能团和/或至少一个脂环族烃官能团和可选的至少一个芳族烃官能团和可选的异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团。
[0169]
[26]根据[22]至[25]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物还包含一个或多个连接链,其中这些连接链中的每一个连接链连接所述多氮丙啶化合物中存在的结构单元a中的两个结构单元a,其中所述连接链优选由2至300个原子组成,更优选由5至250个原子组成,最优选由6至100个原子组成。
[0170]
[27]根据实施方式[26]所述的多氮丙啶化合物,其中在结构单元a中的氨基甲酸酯的n原子与另一个n原子(其存在于连接链中或为另一结构单元a的氨基甲酸酯的n原子)的之间的连续c原子和可选的o原子的数量为至多9。
[0171]
[28]根据实施方式[22]至[27]所述的多氮丙啶化合物,其中所述多氮丙啶化合物是通过使具有以下结构式的化合物b与具有脂肪族反应性的多异氰酸酯反应而获得的:
[0172][0173]
[29]根据实施方式[28]所述的多氮丙啶化合物,其中具有脂肪族反应性的多异氰
酸酯选自1,5-五亚甲基二异氰酸酯pdi、1,6-六亚甲基二异氰酸酯hdi、异佛尔酮二异氰酸酯ipdi、4,4'-二环己基甲烷二异氰酸酯h12mdi、2,2,4-三甲基六亚甲基二异氰酸酯、2,4,4-三甲基六亚甲基二异氰酸酯、四甲基二甲苯二异氰酸酯tmxdi(所有异构体)和更高分子量的变体,如例如它们的异氰脲酸酯或亚氨基噁二嗪二酮。
[0174]
[30]根据实施方式[28]所述的多氮丙啶化合物,其中所述多氮丙啶化合物的连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、芳族烃官能团和脂肪族烃官能团(例如当使用tmxdi制备多氮丙啶化合物时),或者所述连接基团由以下连续官能团的阵列组成:脂环族烃官能团、脂肪族烃官能团和脂环族烃官能团(例如当使用h12mdi制备多氮吡啶化合物时),或者所述连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、异氰脲酸酯官能团或亚氨基噁二嗪二酮官能团和脂肪族烃官能团,或者所述连接基团由以下连续官能团的阵列组成:脂肪族烃官能团、异氰脲酸酯官能团和脂肪族烃官能团(例如当使用1,6-六亚甲基二异氰酸酯的异氰脲酸酯和/或1,5-五亚甲基二异氰酸酯的异氰脲酸酯制备多氮丙啶化合物时)。
[0175]
[31]一种多氮丙啶化合物,所述多氮丙啶化合物具有2至6个、优选2至4个、更优选2至3个如在实施方式[1]中所定义的结构单元(a),其中r1、r2、r3、r4、r'、r
″
和m如在实施方式[1]至[10]中任一项所定义,其中多氮丙啶化合物的分子量为600道尔顿至5000道尔顿,优选至少700道尔顿,更优选至少800道尔顿,甚至更优选至少840道尔顿并且优选至多3800道尔顿,更优选至多3600道尔顿,更优选至多3000道尔顿,更优选至多1600道尔顿,甚至更优选至多1200道尔顿,并且其中所述多氮丙啶化合物是通过使至少多异氰酸酯与具有以下结构式的化合物b反应而获得的:
[0176][0177]
其中化合物b与多异氰酸酯的摩尔比为2至6,更优选2至4,并且最优选2至3,并且其中m、r'、r
″
、r1、r2、r3和r4如实施方式[1]至[10]中任一项所定义。
[0178]
[32]根据实施方式[31]所述的多氮丙啶化合物,其中所述多异氰酸酯为具有脂肪族反应性的多异氰酸酯,优选选自1,5-五亚甲基二异氰酸酯pdi、1,6-六亚甲基二异氰酸酯hdi、异佛尔酮二异氰酸酯ipdi、4,4'-二环己基甲烷二异氰酸酯h12mdi、2,2,4-三甲基六亚甲基二异氰酸酯、2,4,4-三甲基六亚甲基二异氰酸酯、四甲基二甲苯二异氰酸酯tmxdi(所有异构体)和更高分子量的变体,如例如它们的异氰脲酸酯或亚氨基噁二嗪二酮。
[0179]
[33]根据实施方式[31]或[32]所述的多氮丙啶化合物,其中化合物b是通过使至少非oh官能的单环氧化物化合物与具有以下结构式的氮丙啶反应而获得的:
[0180][0181]
其中r1、r2、r3和r4如实施方式[1]或[2]至[7]中任一项中所定义,优选地所述非oh官能的单环氧化物化合物选自由以下项组成的组:环氧乙烷、环氧丙烷、2-乙基环氧乙烷、
正丁基缩水甘油醚、2-乙基己基缩水甘油醚、新癸酸缩水甘油酯以及它们的任何混合物。
[0182]
[34]根据[1]至[33]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物含有聚氧丙烯(-o-chch3-ch2-)
x
基团或聚四氢呋喃(-o-ch2-ch2-ch2-ch2)
x
基团或优选地聚氧乙烯(-o-ch2-ch2-)
x
基团,或者以相对于多氮丙啶化合物的至少0.1重量%,更优选至少6重量%,更优选至少10重量%的量并且优选小于45重量%,更优选小于25重量%并且最优选小于16重量%的量含有聚氧丙烯(-o-chch3-ch2-)
x
基团或聚四氢呋喃(-o-ch2-ch2-ch2-ch2)
x
基团或优选地聚氧乙烯(-o-ch2-ch2-)
x
基团。
[0183]
[35]根据[1]至[34]中的任何实施方式所述的多氮丙啶化合物,其中在如上面所定义的多氮丙啶化合物中m
n
高于2200道尔顿量,优选m
n
高于1600道尔顿的烷氧基聚(乙二醇)(优选甲氧基聚(乙二醇)(mpeg))和/或聚(乙二醇)(peg)链的量优选小于35重量%,更优选小于15重量%,更优选小于5重量%,并且最优选为0重量%,并且在多氮丙啶化合物中存在的甲氧基聚(乙二醇)(mpeg)和/或聚(乙二醇)(peg)链的m
n
优选低于1100道尔顿,更优选低于770道尔顿,最优选低于570道尔顿。
[0184]
[36]根据[1]至[35]中的任何实施方式所述的多氮丙啶化合物,其中所述多氮丙啶化合物中的氨基甲酸酯键的量为至少5重量%,更优选至少5.5重量%,更优选至少6重量%,更优选至少9重量%,更优选至少12重量%,并且优选小于25重量%,优选小于20重量%,并且优选地所述多氮丙啶化合物的氮丙啶当量重量(多氮丙啶的分子量除以氮丙啶基的数量)为至少200道尔顿,更优选至少230道尔顿并且甚至更优选至少260道尔顿,并且优选至多2500道尔顿,更优选至多1000道尔顿并且甚至更优选至多500道尔顿。
[0185]
[37]一种交联剂组合物,所述交联剂组合物包含至少一种根据实施方式[1]至[36]中任一项所述的多氮丙啶化合物,其中所述交联剂组合物中存在的所述根据[1]至[36]中任一项所述的多氮丙啶化合物的分子量在600道尔顿至5000道尔顿的范围内,优选至多3800道尔顿,优选地所述分子量为至多3800道尔顿,更优选至多3600道尔顿,更优选至多3000道尔顿,更优选至多1600道尔顿,甚至更优选至多1200道尔顿,并且所述分子量为优选至少700道尔顿,更优选至少800道尔顿,甚至更优选至少840道尔顿,并且最优选至少1000道尔顿;优选地所述组合物中每个含氮丙啶基的分子中的氮丙啶基的平均数量为至少1.8个,优选至少2个,更优选至少2.2个,并且优选小于10个,更优选小于6个并且最优选小于4个,最优选地所述组合物中每个含氮丙啶基的分子中的氮丙啶基的平均数量为2.2个至3个。
[0186]
[38]根据实施方式[1]至[37]中任一项所述的包含至少一种多氮丙啶化合物的交联剂组合物,其中所述多氮丙啶化合物中存在的氨基甲酸酯键的计算平均量为相对于所述组合物中存在的所述多氮丙啶化合物的总重量的至少5重量%,更优选至少5.5重量%,更优选至少6重量%,更优选至少9重量%,更优选至少12重量%,并且优选小于25重量%,优选小于20重量%。
[0187]
[39]根据实施方式[1]至[38]中任一项所述的包含至少一种多氮丙啶化合物的交联剂组合物,其中相对于所述交联剂组合物的总重量,分子量低于250道尔顿,更优选低于350道尔顿,甚至更优选低于450道尔顿,甚至更优选低于550道尔顿并且甚至更优选低于580道尔顿的氮丙啶官能分子的量低于5重量%,更优选低于2重量%,更优选低于1重量%,更优选低于0.5重量%并且最优选低于0.1重量%,其中所述分子量是如以下实验部分中所
述使用lc-ms测定的。
[0188]
[40]一种包含多氮丙啶的组合物,其中所述多氮丙啶具有以下结构单元(a)中的至少两个结构单元(a):
[0189][0190]
其中
[0191]
r1为h;
[0192]
r2和r4选自h、含有1至8个碳原子并且可选地在链中含有一个或多个杂原子的脂肪(脂环)族基团、苯基、苄基和吡啶基;
[0193]
r3为含有1至8个碳原子并且可选地在链中含有一个或多个杂原子的脂肪(脂环)族基团、苯基、苄基和吡啶基;
[0194]
或者r2和r3(在r2与h不同的情况下)可为同一饱和脂环族基团的组成部分;
[0195]
r'=h或具有1至12个碳原子的烷基;
[0196]
r
″
=h、具有1至12个碳原子的脂肪族基团、具有5至12个碳原子的脂环族基团、具有6至12个碳原子的芳族基团、ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(ocr
″″′
hcr
″″′
h)
n-och3,其中r
″′
和r
″″
独立地为具有1至12个碳原子的烷基,n为1至35,并且r
″″′
独立地为h或具有1至12个碳原子的烷基;
[0197]
或者r'和r
″
可为含有5至8个碳原子的同一饱和脂环族基团的组成部分;
[0198]
m为1至35的整数,
[0199]
其中所述多氮丙啶中存在的氮丙啶基通过至少一条不含(硫)酯官能团并且不含双硫酯官能团并且不含二硫化物官能团的原子链连接,意味着通过该原子链连接的氮丙啶基是通过不是酯基、硫酯基、二硫酯基和二硫化物基的组成部分的键连接的,并且优选也不是(硫代)酰胺基的组成部分、优选也不是缩醛基的组成部分、并且优选也不是磷酸酯(膦酸酯)基的组成部分的键连接的;并且
[0200]
其中所述组合物的数均分子量在550道尔顿至5000道尔顿的范围中。
[0201]
[41]根据实施方式[40]所述的组合物,其中在连接氮丙啶基团的原子链上不存在含有(硫)酯官能团的侧基,具体地,优选在连接氮丙啶基团的原子链上不存在由酸制备的含有(硫)酯官能团的侧基,其中与羰基相邻的碳原子为仲碳原子。
[0202]
[42]根据实施方式[40]或[41]所述的组合物,其中根据本发明的多氮丙啶组合物中存在的多氮丙啶为限定的分子,其中所述多氮丙啶优选含有至少3个所述结构单元(a)并且优选少于10个所述结构单元(a),更优选少于6个所述结构单元(a)并且更优选少于4个所述结构单元(a),最优选地所述多氮丙啶含有3个所述结构单元(a)。
[0203]
[43]根据实施方式[40]至[42]中任一项所述的组合物,其中根据本发明的多氮丙啶组合物为限定的分子的混合物,并且所述多氮丙啶中存在的结构单元(a)的平均量为优选至少2.2个并且优选小于10个,更优选小于6个并且最优选小于4个,优选地所述多氮丙啶中存在的结构单元(a)优选地彼此相同。
[0204]
[44]根据实施方式[40]至[43]中任一项所述的化合物,其中所述多氮丙啶中存在
的氨基甲酸酯键的平均量为至少5重量%,更优选至少5.5重量%,更优选至少6重量%,更优选至少9重量%,更优选至少12重量%,并且优选小于25重量%,优选小于20重量%(相对于所述组合物中存在的多氮丙啶的总量),其中优选使用具有至少2个异氰酸酯基的多异氰酸酯来制备根据本发明的多氮丙啶组合物,所述具有至少2个异氰酸酯基的多异氰酸酯的量为相对于用于制备所述多氮丙啶组合物的含异氰酸酯的化合物的总量的优选>50重量%,更优选>80重量%,最优选>100重量%。
[0205]
[45]根据[40]至[44]中的任何实施方式所述的组合物,其中所述多氮丙啶中存在的氮丙啶基通过至少一个不含(硫代)酰胺官能团、不含缩醛官能团并且不含磷酸酯(膦酸酯)官能团的原子链连接,并且优选地,连接多氮丙啶的两个氮丙啶基团q的原子链优选具有4至50个原子(不包括氮丙啶基团的原子),更优选16至34个原子。
[0206]
[46]根据[40]至[45]中的任何实施方式所述的组合物,其中r4为h,并且r2和r3为同一饱和脂环族基团的组成部分,或者r2为h,r3为ch3并且r4为h或ch3,更优选r4为h。
[0207]
[47]根据[40]至[45]中的任何实施方式所述的组合物,其中m为1至35的整数,优选m为1至6的整数,更优选m为1至4的整数,最优选m=1。
[0208]
[48]根据[40]至[47]中的任何实施方式所述的组合物,其中n为6至20的整数,并且优选地r'为h并且r
″
=具有1至8个碳原子的烷基(优选为ch3或ch2ch3)、ch
2-o-(c=o)-r
″′
或ch
2-o-r
″″
或ch
2-(och2ch2)
n-och3,并且r
″′
为具有3至12个碳原子的烷基,更优选具有3至12个碳原子的支链烷基,并且r
″″
为具有1至12个碳原子的烷基,更优选r
″
为ch
2-o-(c=o)-r
″′
、ch
2-o-r
″″
或ch
2-(och2ch2)
n-och3。
[0209]
[49]根据[40]至[48]中的任何实施方式所述的组合物,其中所述包含多氮丙啶的组合物的数均分子量(和/或所述多氮丙啶组合物中存在的多氮丙啶的数均分子量)在550道尔顿至5000道尔顿的范围内,优选至多3000道尔顿,更优选至多1600道尔顿,甚至更优选至多1200道尔顿,并且优选至少840道尔顿并且最优选至少1000道尔顿,其中所述包含多氮丙啶的组合物的数均分子量为氮丙啶官能团分子和在所述多氮丙啶组合物中存在的多氮丙啶的制备期间获得的可选副产物的数均分子量,并且所述数均分子量是使用maldi-tof质谱法测定的。
[0210]
[50]根据[40]至[49]中的任何实施方式所述的组合物,其中分子量低于550道尔顿,更优选低于700道尔顿,甚至更优选低于840道尔顿的氮丙啶官能分子的量为相对于所述包含多氮丙啶的组合物的总重量的优选低于5重量%,更优选低于2重量%,更优选低于1重量%,更优选低于0.5重量%,最优选低于0.1重量%。
[0211]
[51]根据[40]至[50]中的任何实施方式所述的组合物,其中多氮丙啶中存在的环状结构(除氮丙啶基团以外)的总量优选为0至3个,更优选0至2个,甚至更优选为1或2个,最优选为1个,所述环状结构优选为异氰脲酸酯环。
[0212]
[52]根据[40]至[51]中的任何实施方式所述的组合物,其中所述多氮丙啶是通过使多异氰酸酯与至少一种根据以下结构q-chr'-chr
″-
oh的β-羟基亚烷基氮丙啶反应而获得的,其中q为根据以下结构式
[0213]
[0214]
其中r'、r
″
、r1、r2、r3和r4与实施方式[50]、[54]或[56]中的任一项中相同。
[0215]
[53]根据[40]至[52]中的任何实施方式所述的组合物,其中所述多异氰酸酯含有至少2个异氰酸酯基,优选平均2.5个异氰酸酯基,更优选平均至少2.8个异氰酸酯基;优选地所述多异氰酸酯选自由以下项组成的组:不含环状基团的脂肪族二异氰酸酯的异氰脲酸酯、不含环状基团的脂肪族二异氰酸酯的亚氨基噁二嗪二酮三聚体、缩二脲三聚体以及它们的任何混合物;更优选地,多异氰酸酯为直链(非支链的脂肪族)二异氰酸酯的异氰脲酸酯或亚氨基噁二嗪二酮三聚体;甚至更优选地,所述多异氰酸酯选自由以下项组成的组:1,6-六亚甲基二异氰酸酯的异氰脲酸酯或亚氨基噁二嗪二酮、1,5-五亚甲基二异氰酸酯的异氰脲酸酯以及它们的任何混合物。
[0216]
[54]根据[52]至[53]中的任何实施方式所述的组合物,其中所述β-羟基亚烷基氮丙啶是通过使至少一种非oh官能的单环氧化物化合物与具有结构式的氮丙啶化合物反应而获得的,其中r1、r2、r3和r4如上面所定义。
[0217]
[55]根据[40]至[54]中的任何实施方式所述的组合物,其中所述多氮丙啶组合物中存在的氮丙啶基基团q的平均量为至少1.9个,优选至少2.5个,更优选至少2.7个,更优选至少2.8个,更优选至少2.9个,并且优选小于6.1个,更优选小于5个,并且最优选小于3.5个。
[0218]
[56]根据[40]至[55]中的任何实施方式所述的组合物,其中所述多氮丙啶是通过使至少以下反应物反应而获得的:
[0219]
(i)氮丙啶与至少非oh官能的单环氧化物化合物的加合物,以及
[0220]
(ii)多异氰酸酯,所述多异氰酸酯的量优选为20重量%至67重量%(相对于反应物的总重量),其中所述多氮丙啶组合物的数均分子量为至多5000道尔顿,优选至多3000道尔顿,更优选至多1600道尔顿,更优选至多1200道尔顿并且至少550道尔顿,更优选至少840道尔顿,最优选至少1000道尔顿,并且多分散度优选小于10,更优选小于5,甚至更优选小于2,并且其中所述多异氰酸酯优选为三异氰酸酯,更优选地所述多异氰酸酯选自由以下项组成的组:1,6-六亚甲基二异氰酸酯的异氰脲酸酯、1,5-五亚甲基二异氰酸酯的异氰脲酸酯以及它们的任何混合物;所述氮丙啶优选为丙烯亚胺(cas编号75-55-8)或2,2-二甲基氮丙啶(cas编号2658-24-4),更优选所述氮丙啶为丙烯亚胺。
[0221]
[57]根据实施方式[56]所述的组合物,其中所述非oh官能的单环氧化物选自由以下项组成的组:环氧乙烷、环氧丙烷、2-乙基环氧乙烷、正丁基缩水甘油醚、2-乙基己基缩水甘油醚、新癸酸缩水甘油酯以及它们的任何混合物。
[0222]
[58]根据[40]至[57]中的任何实施方式所述的组合物,其中所述组合物中存在的多氮丙啶含有氧乙烯(-o-ch2-ch2-)基团和/或氧丙烯(-o-chch3-ch2-)基团,所述基团的量为优选至少0.1重量%,更优选至少6重量%,更优选至少10重量%,并且所述基团的量为优选小于45重量%,更优选小于25重量%并且最优选小于16重量%(相对于组合物中存在的多氮丙啶的总量),优选地,所述组合物中存在的多氮丙啶含有氧乙烯(-o-ch2-ch2-)基团,所述基团的量为优选至少0.1重量%,更优选至少6重量%,更优选至少10重量%,并且
所述量优选小于75重量%,更优选小于35重量%并且最优选小于20重量%(相对于组合物中存在的多氮丙啶的总量)。
[0223]
[59]根据[40]至[58]中的任何实施方式所述的组合物,其中在所述多氮丙啶中m
n
高于2200道尔顿,优选m
n
高于1600道尔顿的烷氧基聚(乙二醇)(优选甲氧基聚(乙二醇)(mpeg))和/或聚(乙二醇)(peg)链的量为优选小于35重量%,更优选小于15重量%,更优选小于5重量%,并且最优选0重量%,并且优选地,所述交联剂中存在的甲氧基聚(乙二醇)(mpeg)和/或聚(乙二醇)(peg)链的m
n
优选低于1100道尔顿,更优选低于570道尔顿。
[0224]
[60]根据[40]至[59]中的任何实施方式所述的组合物,其中所述组合物包含小于15重量%,优选小于5重量%,更优选小于1重量%,并且最优选小于0.1重量%的水。
[0225]
[61]根据[40]至[60]中的任何实施方式所述的组合物,其中为其100%形式(无任何稀释剂例如溶剂和增塑剂)的多氮丙啶优选在25℃下的布氏粘度为至少500mpa.s,更优选至少1200mpa.s,更优选至少3000mpa.s,并且在25℃下的布氏粘度为优选至多1000000mpa.s,更优选至多100000mpa.s,更优选至多30000mpa.s,更优选为10000mpa.s并且最优选5000mpa.s,其中所述布氏粘度是根据iso 2555-89测定的。在一个替代实施方式中,用具有锭子s63的brookfield在25℃下在80%固体、20%二甲基甲酰胺(dmf)中测量所述多氮丙啶的粘度;根据该方法测量的粘度优选在300mpa.s至20000mpa.s的范围内,更优选在500mpa.s至12000mpa.s的范围内,并且最优选在700mpa.s至3000mpa.s的范围内。
[0226]
[62]根据[40]至[61]中的任何实施方式所述的组合物,其中所述组合物中存在的多氮丙啶为适于使羧酸官能聚合物交联的交联剂。
[0227]
[63]如在[1]至[62]中的任何实施方式中所定义的组合物用于使溶解和/或分散在水性介质中的羧酸官能聚合物交联的用途。
[0228]
[64]一种多包装体系,所述多包装体系包括第一包装和第二包装,所述第一包装包含溶解和/或分散在水性介质中的羧酸官能聚合物,所述第二包装包含如在[1]至[66]中的任何实施方式中所定义的组合物,其中所述第一包装和所述第二包装是分开储存的。
[0229]
[68]一种涂料组合物,所述涂料组合物是通过就在施加所述涂料组合物之前混合实施方式[64]所述的多包装体系的第一包装和第二包装而获得的,其中所述涂料组合物包含的氮丙啶基基团q和羧酸基团的量使得羧酸基团上的氮丙啶基基团q的化学计量量(sa)为0.1至2.0,更优选0.2至1.5,甚至更优选0.25至0.95,最优选0.3至0.8。
[0230]
[69]一种具有涂层的基材,所述涂层是通过以下方式获得的:(i)将根据实施方式[68]所述的涂料组合物施加到基材上,以及(ii)通过蒸发挥发物来干燥所述涂料组合物。
[0231]
现在通过参考以下实施例来说明本发明。除非另外指明,否则所有份数、百分比和比率均以重量为基础。
[0232]
所使用的组分和缩写:
[0233]
n3600、n3900、n3400和xp2860获自covestro。
[0234]
(
±
)-烯丙基-2,3-环氧丙基醚(烯丙基缩水甘油醚,cas编号106-92-3)获自acros organics(themo fisher scientific的子公司)。
[0235]
正丁基缩水甘油醚(cas编号2426-08-6)获自alfa aesar(themo fisher scientific的子公司)。
fisher scientific的子公司)。
[0257]
ipdi(5-异氰酸根合-1-(异氰酸根合甲基)-1,3,3-三甲基环己烷,i,异佛尔酮二异氰酸酯,cas编号4098-71-9)获自covestro。
[0258]
tmp(1,1,1-三(羟甲基)丙烷,cas编号77-99-6)获自sigma-aldrich(merck kgaa的子公司)。
[0259]
tdi(甲苯二异氰酸酯,cas编号26471-62-5,t80,2,4-甲苯二异氰酸酯与2,6-甲苯二异氰酸酯的80/20混合物)获自covestro。
[0260]
durez-ter s105-110(基于己二酸和己二醇的oh值为110mg koh/g的聚酯多元醇)获自sumitomo bakelite。
[0261]
pthf650(oh值为172mg koh/g的聚四亚甲基醚二醇)获自basf。
[0262]
新癸酸铋(cas编号34364-26-6)获自tib chemicals ag(mannheim,germany)。
[0263]
吩噻嗪(cas编号92-84-2)获自sigma-aldrich(merck kgaa的子公司)。
[0264]
1-丁醇(cas编号71-36-3)获自sigma-aldrich(merck kgaa的子公司)。
[0265]
1-甲基-2-丙醇乙酸酯(丙二醇甲醚乙酸酯,cas编号108-65-6)获自shell chemicals。
[0266]
肼(16%水溶液,cas编号302-01-2)获自honeywell。
[0267]
二羟甲基丙酸(dmpa,cas编号4767-03-7)获自perstop polyols。
[0268]
三乙胺(tea,cas编号121-44-8)获自arkema。
[0269]
十二烷基硫酸钠(30%水溶液,cas编号73296-89-6)获自basf。
[0270]
甲基丙烯酸甲酯(cas编号80-62-6)获自lucite int。
[0271]
丙烯酸正丁酯(cas编号141-32-2)获自dow chemical。
[0272]
甲基丙烯酸(cas编号79-41-4)获自lucite int。
[0273]
过硫酸铵(cas编号7727-54-0)获自united initiators。
[0274]
氨(25%水溶液,cas编号1336-21-6)获自merck。
[0275]
二丙二醇二甲醚(cas编号34590-94-8)获自dow chemical。
[0276]
2-甲基-1,3-丙二醇(cas编号2163-42-0)获自lyondell。
[0277]
1,4-丁二醇(cas编号110-63-4)获自basf。
[0278]
己二酸(cas编号124-04-9)获自basf。
[0279]
lc-ms
[0280]
使用以下工序进行低分子量级分的lc-ms分析:
[0281]
重量分析地制备材料的约100mg/kg的甲醇溶液,并进行搅拌。将0.5μl的该溶液注入配备有esi-tof-ms检测的uplc中。所使用的柱是在40℃下操作的100
×
2.1mm,1.8um,waters hss t3 c18。流率为0.5ml.min-1。所使用的溶剂为10mm nh4ch3coo(用nh3设置至ph 9.0,洗脱液a)、乙腈(b)和thf(c)。施加在10分钟内从80/20a/b至1/99a/b和在5分钟内从1/99a/b至1/49/50a/b/c的2个二元梯度,在此之后施加起始条件(80/20a/b)。假设所有组分在所有响应范围内均具有直链ms响应,并且所有组分的电离效率相等,对总离子电流信号进行积分。在共洗脱的情况下,对该特定物种的提取离子色谱图进行积分。
[0282]
maldi-tof-ms
[0283]
所有maldi-tof-ms光谱均使用bruker ultraflextreme maldi-tof质谱仪获取。
该仪器配备有在1064nm发射的nd:yag激光器和碰撞室(不用于这些样品)。使用反射器,使用提供准确质量的最高分辨率模式(范围为60-7000m/z)以正离子模式获取光谱。使用三碘化铯(范围为0.3-3.5kda)进行质量校准(校准方法:iav分子表征,代码mc-ms-05)。激光能量为20%。将样品以约50mg/ml溶于thf中。所使用的基质为:dctb(反式2-[3-(4-叔丁基苯基)-2-甲基-2-亚丙烯基]丙二腈),cas编号300364-84-5。通过将20mg溶于1ml thf中来制备基质溶液。除了将三氟乙酸钾(ktfa,cas编号:2923-16-2)用作盐的对比例4、6和8之外,将碘化钠用作盐(nai,cas编号7681-82-5);将10mg溶于1ml thf中,并滴加meoh。样品:基质:盐的比率=10:200:10(μl),在混合后,将0.5μl点样到maldi板上并风干。所报告的信号为理论上以最大量存在于组合物中的多氮丙啶化合物的计算质量的0.5da内的主要峰。在所有情况下,所报告的峰均为所测量的离子的钠或钾加合物。
[0284]
遗传毒性测试
[0285]
通过测定(toxys,leiden,the netherlands)评估实施例和对比例的遗传毒性。toxtracker测定是一组若干经验证的基于绿色荧光蛋白(gfp)的小鼠胚胎干(mes)报道基因细胞系,所述报道基因细胞系可用于在单次测试中鉴定新开发的化合物的生物活性和潜在的致癌特性。该方法使用两步法。
[0286]
在第一步骤中,使用野生型mes细胞(品系b4418)进行行剂量范围发现。测试每种化合物的20种不同浓度,以dmso中的10mm作为最高浓度开始,以及19个连续2倍稀释。
[0287]
接下来,使用用于检测dna损伤的与报道基因连锁的特定基因来评估实施例和对比例的遗传毒性;所述特定基因即bscl2(如由us9695481b2和ep2616484b1阐明的)和rtkn(hendriks等人,toxicol.sci.2015,150,190-203)生物标记物。在不存在和存在基于大鼠s9肝提取物的代谢系统(aroclor1254诱导的大鼠,moltox,boone,nc,usa)的情况下,在10%、25%和50%的细胞毒性下评估遗传毒性。将独立的细胞系接种到96孔细胞培养板中,在细胞接种到96孔板中后24小时处将含有稀释的测试物质的新鲜es细胞培养基添加到细胞中。对于每种测试的化合物,以2倍稀释度测试五种浓度。最高样品浓度将诱导明显的细胞毒性(50-70%)。在无或低细胞毒性的情况下,将10mm或最大可溶混合物浓度用作最大测试浓度。通过在暴露24小时后使用guava easycyte 10ht流式细胞仪(millipore)进行细胞计数来测定细胞毒性。
[0288]
总是将gfp报告基因诱导与媒介物对照处理进行比较。对于特定化合物,所有孔中的dmso浓度均相似并且从未超过1%。在至少三个完全独立的重复实验中测试所有化合物。所有实验中均包括使用顺铂的阳性对照处理(dna损伤)。通过添加s9肝提取物评估代谢性。在s9和所需的辅因子(regensysa+b,moltox,boone,nc,usa)存在下,将细胞暴露于五种浓度的测试化合物3小时。在洗涤后,将细胞在新鲜的es细胞培养基中孵育24小时。在暴露24小时后,使用guava easycyte 10ht流式细胞仪(millipore)测定gfp报告基因的诱导。仅确定完整单个细胞中的gfp表达。测量每个孔中的平均gfp荧光和细胞浓度,将所述平均gfp荧光和细胞浓度用于细胞毒性评定。使用toxplot软件(toxys,leiden,the netherlands)分析数据。所报道的诱导水平处于在s9大鼠肝提取物存在下暴露3小时并恢复24小时后或替代地在不存在s9大鼠肝提取物的情况下暴露24小时后诱导10%、25%和50%的细胞毒性的化合物浓度下。
[0289]
生物标记物的阳性诱导水平被定义为等于或高于在不存在或存在代谢系统大鼠
s9肝提取物的情况下,10%、25%和50%细胞毒性中的至少一者处的2倍诱导;弱阳性诱导被定义为在不存在或存在代谢系统大鼠s9肝提取物的情况下,10%、25%和50%细胞毒性中的至少一者处的高于1.5倍和低于2倍诱导(但在10%、25%和50%细胞毒性处低于2倍),并且阴性诱导被定义为在不存在或存在基于大鼠s9肝提取物的代谢系统的情况下,低于或等于10%、25%和50%细胞毒性处的1.5倍诱导。
[0290]
对比例1
[0291]
对比例1为cx-100,三羟甲基丙烷三(2-甲基-1-氮丙啶丙酸酯)。
[0292]
化学结构如下所示。
[0293][0294]
作为参考,基于来自din 68861-1标准的工序,使用涂层表面上的斑点测试来评定三羟甲基丙烷三(2-甲基-1-氮丙啶丙酸酯)作为交联剂的性能。对于这些测试,将0.23份化合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.56份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试c1-1)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉(cotton wool)浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0295]
乙醇斑点测试
[0296][0297]
遗传毒性测试
[0298][0299]
对比例2
[0300]
将508.7克agisyn 2844和0.26克吩噻嗪装入配备有恒温器的不锈钢反应器中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至35℃,随后在30分钟内添
加25.3克丙烯亚胺。然后将混合物进一步加热至45℃并在该温度下保持36小时。将主组分的计算分子量为800.48da的所得混合物排出,并测试遗传毒性。化学结构如下所示。
[0301][0302]
遗传毒性测试
[0303][0304]
对比例3
[0305]
将10.0克1,3-双(1-异氰酸根合-1-甲基乙基)苯、0.02克新癸酸铋、3.56克1-(2-羟乙基)亚乙基亚胺、1.83克三羟甲基丙烷和87克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至50℃,在该温度下保持15分钟,然后进一步加热至75℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得淡黄色蜡。理论主组分的计算分子量为1127.66da(三个氮丙啶)和418.26da(两个氮丙啶),化学结构如下所示。
[0306][0307]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1150.66da;观察到的[m+na+]=1150.56da。
[0308][0309]
通过maldi-tof-ms确认分子量:计算的[m+na+]=441.26da;观察到的[m+na+]=441.20da。
[0310]
遗传毒性测试
[0311][0312]
对比例4
[0313]
将15.0克desmodur n 3600和75克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至50℃,在此之后添加6.80克1-(2-羟乙基)亚乙基亚胺。在15分钟后,将0.03克新癸酸铋装入反应烧瓶中,然后将所述反应烧瓶进一步加热至60℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的略带黄色的高粘度液体。理论主组分的计算分子量为765.47da,化学结构如下所示。
[0314][0315]
通过maldi-tof-ms确认分子量:计算的[m+k+]=804.43da;观察的[m+k+]=804.27da。
[0316]
遗传毒性测试
[0317][0318]
对比例5
[0319]
将2.60克1-(吖丙啶-1-基)丙-2-醇、0.02克新癸酸铋和32克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在15分钟内将5.00克desmodur n 3600在32克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的高粘度液体。理论主组分的计算分子量为807.52da,化学结构如下所示。
[0320][0321]
通过maldi-tof-ms确认分子量:计算的[m+na+]=830.52da;观察到的[m+na+]=830.47da。
[0322]
遗传毒性测试
[0323][0324]
对比例6
[0325]
将9.00克1,6-己烷二异氰酸酯和74克2-甲基四氢呋喃装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至40℃,在达到该温度时,逐渐装入12.20克的1-(2-甲基吖丙啶-1-基)丙-2-醇。在急剧放热后,将反应温度稳定在60℃并保持在该温度。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的粘性液体。理论主组分的计算分子量为398.29da,化学结构如下所示。
[0326][0327]
通过maldi-tof-ms确认分子量:计算的[m+k+]=437.25da;观察的[m+k+]=437.20da。
[0328]
遗传毒性测试
[0329][0330]
对比例7
[0331]
将3.00克ipdi、28克dmf和3.08克1-(2-甲基吖丙啶-1-基)丙-2-醇装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热到50℃,在达到该温度时,添加0.02克新癸酸铋。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的高粘度液体。理论主组分的计算分子量为452.34da,化学结构如下所示。
[0332][0333]
通过maldi-tof-ms确认分子量:计算的[m+na+]=475.34da;观察到的[m+na+]=475.32da。
[0334]
遗传毒性测试
[0335][0336]
对比例8
[0337]
将8.95克1-(2-甲基吖丙啶-1-基)丙-2-醇、0.02克新癸酸铋和54克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将10.0克1,3-双(1-异氰酸根合-1-甲基乙基)苯在54克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的粘性液体。理论主组分的计算分子量为474.32da,化学结构如下所示。
[0338][0339]
通过maldi-tof-ms确认分子量:计算的[m+k+]=513.28da;观察的[m+k+]=513.19da。
[0340]
遗传毒性测试
[0341][0342]
对比例9
[0343]
将3.00克h12mdi、27克dmf和2.61克1-(2-甲基吖丙啶-1-基)丙-2-醇装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热到50℃,在达到该温度时,添加0.02克新癸酸铋。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的高粘度液体。理论主组分的计算分子量为492.37da,化学结构如下所示。
[0344][0345]
通过maldi-tof-ms确认分子量:计算的[m+na+]=515.37da;观察到的[m+na+]=515.35da。
[0346]
遗传毒性测试
[0347][0348]
实施例1
[0349]
将20.0克desmodur n 3600、11.98克1-(2-甲基吖丙啶-1-基)丙-2-醇和106克2-甲基四氢呋喃装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至50℃,在该温度下保持15分钟,然后进一步加热至60℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的高粘度液体。理论主组分的计算分子量为849.57da,化学结构如下所示。
[0350][0351]
通过maldi-tof-ms确认分子量:计算的[m+na+]=872.57da;观察到的[m+na+]=872.53da。通过lc-ms测定并定量质量低于580da的以下组分:
[0352][0353]
在组合物中以0.06重量%存在。
[0354]
遗传毒性测试
[0355][0356]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.41份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.67份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试1-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试1-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0357]
乙醇斑点测试
[0358][0359]
使用在具有不同粘合剂体系的涂料表面上的斑点测试来评定合成的化合物作为交联剂的性能。
[0360]
水性聚氨酯粘合剂如下合成。
[0361]
在配备有温度计和顶置式搅拌器的1l烧瓶中装入dmpa(13.4克)、pthf650(166.1克)和ipdi(156.5克)。将反应混合物置于n2气氛下,加热至50℃,并添加0.03g新癸酸铋。使混合物放热,并在90℃保持2.5小时。所得氨基甲酸酯预聚物的nco含量为按固体计8.00%(理论上8.80%)。将预聚物冷却至75℃并添加tea(9.12克),并将所得混合物搅拌15分钟。通过在30分钟内在室温下将320.2克该预聚物进料到脱矿质水(700克)中来制备所得预聚物的分散体。在进料完成后,将混合物搅拌5分钟并添加肼(16%水溶液,61.6克)。将分散体搅拌另外1小时。
[0362]
为了进一步的斑点测试,将0.35份的交联剂组合物与0.07份的二丙二醇甲醚混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份上述水性聚氨酯粘合剂中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试1-3)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试1-4)。将膜在25℃下干燥1小时,然后在50℃下退火16小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0363]
乙醇斑点测试
[0364][0365]
水性丙烯酸粘合剂如下合成。
[0366]
在配备有温度计和顶置式搅拌器的2l四颈烧瓶中装入十二烷基硫酸钠(水溶液中30%的固体,18.6克的溶液)和脱矿质水(711克)。将反应器相置于n2气氛下并加热至82℃。将脱矿质水(112克)、十二烷基硫酸钠(水中30%的固体,37.2克的溶液)、甲基丙烯酸甲酯(174.41克)、丙烯酸正丁酯(488.44克)和甲基丙烯酸(34.88克)的混合物放入大进料漏斗中,并用顶置式搅拌器进行乳化(单体进料)。将过硫酸铵(1.75克)溶解在脱矿质水(89.61克)中,并放入小进料漏斗中(引发剂进料)。将过硫酸铵(1.75克)溶解在脱矿质水(10.5克)中,并将该溶液添加到反应器相中。之后立即将5体积%的单体进料添加至反应器相中。然后使反应混合物放热至85℃,并在85℃保持5分钟。然后,在90分钟内将残留的单体进料和引发剂进料进料到反应混合物中,保持在85℃的温度下。在进料完成后,将单体进料漏斗用脱矿质水(18.9克)冲洗,并将反应温度保持在85℃下45分钟。随后,将混合物冷却至室温,并用氨溶液(在脱矿质水中6.25重量%)调至ph=7.2,并用另外的脱矿质水调至40%固体。
[0367]
为了进一步的斑点测试,将0.89份的交联剂组合物与0.18份的二丙二醇甲醚混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份上述水性聚丙烯酸酯粘合剂中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试1-5)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试1-6)。将膜在25℃下干燥1小时,然后在50℃下退火16小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0368]
乙醇斑点测试
[0369][0370]
聚酯粘合剂如下合成。
[0371]
在配备有蒸馏装置、温度计和顶置式搅拌器的2l反应器中装入2-甲基-1,3-丙二醇(151.9克)、1,4-丁二醇(152.1克)、己二酸(446.3克)和二聚脂肪酸(371.2克)。将反应混合物加热至220℃,并通过蒸馏去除反应产生的水。减压除去剩余的水,直到混合物的酸值小于40mg koh/g。为了进一步的斑点测试,将42份聚酯粘合剂与8.4份甲基乙基酮(聚酯混合物)混合。
[0372]
为了进一步的斑点测试,将1.93份的交联剂组合物与0.39份的二丙二醇甲醚混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至25.4份上述聚酯混合物中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用
100μm线材涂布器施加到leneta测试卡上(测试1-7)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试1-8)。
[0373]
乙醇斑点测试
[0374][0375]
实施例2
[0376]
将5.93克1-(2-甲基吖丙啶-1-基)丙-2-醇、0.02克新癸酸铋和40.18克2-甲基四氢呋喃装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将10.0克desmodur n 3900在40.18克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至75℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得淡黄色的高粘度液体。理论主组分的计算分子量为849.57da,化学结构如下所示。
[0377][0378]
通过maldi-tof-ms确认分子量:计算的[m+na+]=872.57da;观察到的[m+na+]=872.62da。通过lc-ms测定并定量质量低于580da的以下组分:
[0379][0380]
在组合物中以1.4重量%存在。
[0381]
遗传毒性测试
[0382][0383]
实施例3
[0384]
将2.14克1-(2-甲基吖丙啶-1-基)丙-2-醇、2.72克平均mn为350da的聚(乙二醇)
单甲醚,0.02克新癸酸铋和28克2-甲基四氢呋喃装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将5.13克desmodur n3600在28克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的高粘度液体。理论主组分的计算分子量为849.57da(三个氮丙啶)、1074.68da(两个氮丙啶,7eg重复单元)、1118.70da(两个氮丙啶,8eg重复单元)和1162.73da(两个氮丙啶,9eg重复单元),化学结构如下所示。
[0385][0386]
通过maldi-tof-ms确认分子量:计算的[m+na+]=872.57da;观察到的[m+na+]=872.53da。
[0387][0388]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1097.68da;观察到的[m+na+]=1097.63da。
[0389][0390]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1141.70da;观察到的[m+na+]=1141.66da。
[0391][0392]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1185.73da;观察到的[m+na+]=1185.68da。
[0393]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.71份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.87份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试3-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试3-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0394]
乙醇斑点测试
[0395][0396]
遗传毒性测试
[0397][0398][0399]
实施例4
[0400]
将15.0克desmodur n 3600、7.09克1-(2-甲基吖丙啶-1-基)丙-2-醇、8.21克平均mn为500da的聚(乙二醇)单甲醚和110克2-甲基四氢呋喃装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至50℃,在该温度下保持15分钟,然后进一步加热至60℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的高
粘度液体。理论主组分的计算分子量为849.57da(三个氮丙啶)、1250.78da(两个氮丙啶,11eg重复单元)、1294.81da(两个氮丙啶,12eg重复单元)和1338.84da(两个氮丙啶,13eg重复单元),化学结构如下所示。
[0401][0402]
通过maldi-tof-ms确认分子量:计算的[m+na+]=872.57da;观察到的[m+na+]=872.54da。
[0403][0404]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1273.78da;观察到的[m+na+]=1273.76da。
[0405][0406]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1317.81da;观察到的[m+na+]=1317.78da。
[0407][0408]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1361.84da;观察到的[m+na+]=1361.81da。通过lc-ms测定并定量质量低于580da的以下组分:
[0409][0410]
在组合物中以0.26重量%存在。
[0411]
遗传毒性测试
[0412][0413]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.66份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.84份所得溶液添加至21份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试4-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试4-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0414]
乙醇斑点测试
[0415]
[0416]
使用在具有不同粘合剂体系的涂料表面上的斑点测试来评定合成的化合物作为交联剂的性能。
[0417]
为了进一步的斑点测试,将0.55份的交联剂组合物与0.11份的二丙二醇甲醚混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份如在实施例1中所述制备的水性聚氨酯粘合剂中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试4-3)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试4-4)。将膜在25℃下干燥1小时,然后在50℃下退火16小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0418]
乙醇斑点测试
[0419][0420]
为了进一步的斑点测试,将1.41份的交联剂组合物与0.28份的二丙二醇甲醚混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份如在实施例1中所述制备的水性聚丙烯酸酯粘合剂中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试4-5)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试4-6)。将膜在25℃下干燥1小时,然后在50℃下退火16小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0421]
乙醇斑点测试
[0422][0423]
在另一测试中,将3.07份的交联剂组合物与0.61份的二丙二醇甲醚混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至25.4份如在上面的实施例1所述制备的聚酯混合物中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试4-7)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试4-8)。
[0424]
将所有膜在25℃下干燥1小时,然后在50℃下退火16小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0425]
乙醇斑点测试
[0426][0427]
实施例5
[0428]
将15.0克desmodur n 3600、7.09克1-(2-甲基吖丙啶-1-基)丙-2-醇、8.21克平均mn为1000da的聚(乙二醇)单甲醚和112克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将混合物加热至50℃,添加0.03克二月桂酸二丁基锡,并且在15分钟后,将混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的高粘度液体。理论主组分的计算分子量为849.57da(三个氮丙啶)和1735.07da(两个氮丙啶,22eg重复单元),化学结构如下所示。
[0429][0430]
遗传毒性测试
[0431][0432]
实施例6
[0433]
将6.00克desmodur n 3600和89克2-甲基四氢呋喃装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌。然后将该混合物加热至50℃,在达到该温度时,将2.84克1-(2-甲基吖丙啶-1-基)丙-2-醇、13.14克平均mn为2000da的聚(乙二醇)单甲醚和0.02克的新癸酸铋装入反应烧瓶中,然后进一步加热至60℃。以规律间隔取样,并使
用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得白色固体蜡。理论主组分的计算分子量为849.57da(三个氮丙啶)和2747.67da(两个氮丙啶,45eg重复单元),化学结构如下所示。
[0434][0435]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将1.22份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将1.21份所得溶液添加至21份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试6-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试6-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0436]
乙醇斑点测试
[0437][0438]
遗传毒性测试
[0439][0440]
实施例7
[0441]
将10ml反应小瓶置于n2气氛下,装入丙烯亚胺(2.28克)、1,2-环氧丁烷(3.40克),加盖,加热至55℃,在此之后将混合物在t=55℃下搅拌4天。真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0442]
将1.62克desmodur n 3600、0.02克新癸酸铋和8.20克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在15分钟内将1.04克来自第一步骤的产物在8.20克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将8.20克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.05克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。真空蒸发溶剂至30%固体,得到澄清液体。理论主组分的计算分子量为891.62da,化学结构如下所示。
[0443][0444]
通过maldi-tof-ms确认分子量:计算的[m+na+]=914.62da;观察到的[m+na+]=914.66da。通过lc-ms测定并定量质量低于580da的以下组分:
[0445][0446]
在组合物中以0.09重量%存在。
[0447]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,在连续搅拌下将1.56份组合物(即在二甲基甲酰胺中30%固体时)添加到15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试7-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试7-2)。将膜在25℃下干燥16小时,然后在
50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0448]
乙醇斑点测试
[0449][0450]
遗传毒性测试
[0451][0452]
实施例8
[0453]
向20ml反应小瓶中装入丙烯亚胺(2.95克)、反式2,3-环氧丁烷(3.75克)和k2co3(1.0克),加盖,加热至55℃,然后将混合物在t=55℃下搅拌11天。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0454]
将0.96克desmodur n 3600、0.02克新癸酸铋和4.90克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在15分钟内将0.62克来自第一步骤的产物在4.90克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将4.90克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.03克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。真空蒸发溶剂至45%固体,得到低粘度的粉红色液体。理论主组分的计算分子量为891.62da,化学结构如下所示。
[0455][0456]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,在连续搅拌下将1.01份组合物(即在二甲基甲酰胺中45%固体时)添加到15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,
将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试8-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试8-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0457]
乙醇斑点测试
[0458][0459]
遗传毒性测试
[0460][0461]
实施例9
[0462]
向反应小瓶中装入丙烯亚胺(3.53克)、烯丙基缩水甘油醚(5.07克)和k2co3(0.5克),加盖并加热至80℃,然后将混合物在t=80℃下搅拌20小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0463]
将4.0克desmodur n 3600、0.02克新癸酸铋和7.50克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在10分钟内将3.41克来自第一步骤的产物在7.50克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将7.50克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.13克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。所得溶液为澄清液体。理论主组分的计算分子量为1017.65da,化学结构如下所示。
[0464][0465]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1040.64da;观察到的[m+na+]=1040.75da。通过lc-ms测定并定量质量低于580da的以下组分:
[0466][0467]
在组合物中以0.098重量%存在,并且
[0468][0469]
在组合物中以小于0.01重量%存在。
[0470]
遗传毒性测试
[0471][0472]
实施例10
[0473]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0474]
将46.54克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)和28.63克1-(2-甲基吖丙啶-1-基)丙-2-醇与0.02克新癸酸铋和32.54克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将100克desmodur n 3600在32.54克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,再将10克2-甲基四氢呋喃通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得淡黄色的高粘度液体。理论主组分的计算分子量为849.57da(三个甲基侧基)、921.63da(两个甲基侧基、一个丁氧甲基侧基)、993.68da(一个甲基侧基、两个丁氧甲基侧基)和1065.74da(三个丁氧甲基侧基),化学结构如下所示。
[0475][0476]
通过maldi-tof-ms确认分子量:计算的[m+na+]=872.57da;观察到的[m+na+]=872.59da。
[0477][0478]
通过maldi-tof-ms确认分子量:计算的[m+na+]=944.63da;观察到的[m+na+]=944.66da。
[0479][0480]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1016.68da;观察到的[m+na+]=1016.72da。
[0481][0482]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1088.74da;观察到的[m+na+]=1088.79da。
[0483]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.74份组合物与0.74份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试10-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试10-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0484]
乙醇斑点测试
[0485][0486]
遗传毒性测试
[0487][0488]
对比例10
[0489]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0490]
将20.9克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸铋一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在30分钟内将10.0克hdi滴加到该反应烧瓶中,然后将该混合物进一步加热到80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程。在3小时后,将0.25g 1-丁醇添加至反应混合物中,并将混合物在80℃下搅拌1小时。在冷却至室温后,获得了淡黄色的高粘度液体。理论主组分的计算分子量为542.40da,化学结构如下所示。
[0491][0492]
通过maldi-tof-ms确认分子量:计算的[m+na+]=565.40da;观察到的[m+na+]=565.49da。
[0493]
遗传毒性测试
[0494][0495]
对比例11
[0496]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0497]
将20.2克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸
铋一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在30分钟内将10.0克tdi滴加到该反应烧瓶中,然后将该混合物进一步加热到80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。在冷却至室温后,获得不透明的固体。理论主组分的计算分子量为548.36da,化学结构如下所示。
[0498][0499]
通过maldi-tof-ms确认分子量:计算的[m+na+]=571.36da;观察到的[m+na+]=571.42da。
[0500]
遗传毒性测试
[0501][0502]
实施例11
[0503]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0504]
将8.72克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸铋和24.85克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将6.0克1,3-双(1-异氰酸根合-1-甲基乙基)苯在24.85克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.18克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。真空去除溶剂,以获得深黄色的高粘度半透明液体。理论主组分的计算分子量为618.44da,化学结构如下所示。
[0505][0506]
通过maldi-tof-ms确认分子量:计算的[m+na+]=641.44da;观察到的[m+na+]=641.44da。通过lc-ms测定并定量质量低于580da的以下组分:
[0507][0508]
在组合物中以0.2重量%存在。
[0509]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.16份组合物与0.16份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至5份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试11-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试11-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0510]
乙醇斑点测试
[0511][0512][0513]
遗传毒性测试
[0514][0515]
实施例12
[0516]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t
=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0517]
将8.12克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸铋和23.62克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将6.0克4,4'-亚甲基双(环己基异氰酸酯)在23.62克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.17克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。真空去除溶剂,以获得淡黄色的高粘度半透明液体。理论主组分的计算分子量为636.48da,化学结构如下所示。
[0518][0519]
通过maldi-tof-ms确认分子量:计算的[m+na+]=659.48da;观察到的[m+na+]=659.47da。通过lc-ms测定并定量质量低于580da的以下组分:
[0520][0521]
在组合物中以0.04重量%存在。
[0522]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.16份组合物与0.16份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至5份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试12-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试12-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0523]
乙醇斑点测试
[0524][0525]
遗传毒性测试
[0526][0527]
实施例13
[0528]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0529]
将20克desmodur n 3400和0.02克新癸酸铋装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在10分钟内将17.88克来自第一步骤的产物滴加到反应烧瓶中,然后将该混合物进一步加热到70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.16克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。产物是淡黄色的高粘度液体。理论主组分的计算分子量为710.49da,化学结构如下所示。
[0530][0531]
通过maldi-tof-ms确认分子量:计算的[m+na+]=733.49da;观察到的[m+na+]=733.57da。通过lc-ms测定并定量质量低于580da的以下组分:
[0532][0533]
在组合物中以0.2重量%存在,并且
[0534][0535]
以小于0.01重量%存在。
[0536]
遗传毒性测试
[0537][0538]
实施例14
[0539]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0540]
将1.92克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸铋和19克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将2.00克desmodur n 3600在19克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的淡黄色高粘度液体。理论主组分的计算分子量为1065.74da,化学结构如下所示。
[0541][0542]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1088.74da;观察到的[m+na+]=1088.76da。通过lc-ms测定并定量质量低于580da的以下组分:
[0543][0544]
在组合物中以0.36重量%存在,并且
[0545][0546]
以小于0.01重量%存在。
[0547]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.58份的组合物与0.60份proglyde
tm
dmm(二丙二醇
二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.79份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试14-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试14-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0548]
乙醇斑点测试
[0549][0550]
遗传毒性测试
[0551][0552][0553]
实施例15
[0554]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0555]
将26.98克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸铋和6.79克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将28.00克desmodur n 3900在6.79克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,再将10克2-甲基四氢呋喃通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的淡黄色高粘度液体。理论主组分的计算分子量为1065.74da,化学结构如下所示。
[0556][0557]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1088.74da;观察到的[m+na+]=1088.81da。通过lc-ms测定并定量质量低于580da的以下组分:
[0558][0559]
在组合物中以0.30重量%存在,并且
[0560][0561]
以0.02重量%存在。
[0562]
遗传毒性测试
[0563][0564]
实施例16
[0565]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0566]
将20克desmodur xp 2860和0.02克新癸酸铋装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在10分钟内将16.40克来自第一步骤的产物滴加到反应烧瓶中,然后将该混合物进一步加热到70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.16克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。产物是淡黄色的半透明高粘度液体。理论主组分的计算分子量为770.55da(丙基侧基)、784.57da(丁基侧基)和798.58da(戊基侧基),化学结构如下所示。
[0567][0568]
通过maldi-tof-ms确认分子量:计算的[m+na+]=793.55da;观察到的[m+na+]=793.57da。
[0569][0570]
通过maldi-tof-ms确认分子量:计算的[m+na+]=807.57da;观察到的[m+na+]=807.61da。
[0571][0572]
通过maldi-tof-ms确认分子量:计算的[m+na+]=821.58da;观察到的[m+na+]=821.63da。通过lc-ms测定并定量质量低于580da的以下组分:
[0573][0574]
在组合物中以0.66重量%存在,并且
[0575][0576]
以0.14重量%存在。
[0577]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.62份组合物与0.62份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并
使用100μm线材涂布器施加到leneta测试卡上(测试16-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试16-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0578]
乙醇斑点测试
[0579][0580]
遗传毒性测试
[0581][0582]
实施例17
[0583]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0584]
将18.21克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)和9.45克平均mn为350da的聚(乙二醇)单甲醚与0.02克新癸酸铋和6.29克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在25分钟内将25.0克desmodur n 3600在6.29克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,再将10克2-甲基四氢呋喃通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得淡黄色的高粘度半透明液体。理论主组分的计算分子量为1065.74da(三个氮丙啶)、1218.79da(两个氮丙啶,7eg重复单元)、1262.82da(两个氮丙啶,8eg重复单元)和1306.85da(两个氮丙啶,9eg重复单元),化学结构如下所示。
[0585][0586]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1088.74da;观察到的[m+na+]=1088.90da。
[0587][0588]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1241.79da;观察到的[m+na+]=1241.96da。
[0589][0590]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1285.82da;观察到的[m+na+]=1286.00da。
[0591][0592]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1329.85da;观察到的[m+na+]=1330.03da。通过lc-ms测定并定量质量低于580da的以下组分:
[0593][0594]
在组合物中以0.03重量%存在,并且
[0595][0596]
以小于0.01重量%存在。
[0597]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.98份组合物与0.25份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试17-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试17-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0598]
乙醇斑点测试
[0599][0600]
遗传毒性测试
[0601]
[0602]
实施例18
[0603]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(80.0克)、正丁基缩水甘油醚(126.0克)和k2co3(10.00克)并在30分钟内加热至80℃,在此之后将混合物在t=80℃下搅拌21小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0604]
将49.5克所得物质(1-丁氧基-3-(2-甲基吖丙啶-1-基)丙-2-醇)与35.2克平均mn为500da的聚(乙二醇)单甲醚、0.2克新癸酸铋和425克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将65.2克desmodur n 3600在425克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至75℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的深黄色高粘度液体。理论主组分的计算分子量为1065.74da(三个氮丙啶)、1394.90da(两个氮丙啶,11eg重复单元)、1438.92da(两个氮丙啶,12eg重复单元)和1482.95da(两个氮丙啶,13eg重复单元),化学结构如下所示。
[0605][0606]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1088.74da;观察到的[m+na+]=1088.67da。
[0607][0608]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1417.90da;观察到的[m+na+]=1417.81da。
[0609][0610]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1461.92da;观察到的[m+na+]=1461.84da。
[0611][0612]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1505.95da;观察到的[m+na+]=1505.86da。通过lc-ms测定并定量质量低于580da的以下组分:
[0613][0614]
在组合物中以0.04重量%存在,并且
[0615][0616]
以0.05重量%存在。
[0617]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.83份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.95份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试18-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试18-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0618]
乙醇斑点测试
[0619][0620]
遗传毒性测试
[0621][0622]
出于证实目的,还根据以下指南,使用已建立的ames测试(细菌回变测定)评定交联剂组合物的诱变性:
[0623]
·
oecd指南471。遗传毒理学:细菌反向突变测试。(在1997年7月21日通过)。
[0624]
·
ec指南号440/2008。b部分:确定毒性和其他健康影响的方法,指南b.13/14:“诱变性:使用细菌的反向突变测试”,欧盟官方公报第l142号,2008年5月31日。
[0625]
该测试表明如上所述的交联剂组合物不是诱变性的。
[0626]
实施例19
[0627]
向20ml反应小瓶中装入丙烯亚胺(2.97克)、氧化环己烯(4.06克),加盖,加热至55℃,在此之后将混合物在t=55℃下搅拌8天。真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到白色粉末。
[0628]
将2.00克desmodur n 3600、0.02克新癸酸铋和3.60克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在10分钟内将1.54克来自第一步骤的产物在3.60克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将3.60克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.06克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。产物是稍微不透明的液体。理论主组分的计算分子量为969.66da,化学结构如下所示。
[0629][0630]
通过maldi-tof-ms确认分子量:计算的[m+na+]=992.66da;观察到的[m+na+]=992.77da。通过lc-ms测定并定量质量低于580da的以下组分:
[0631][0632]
在组合物中以0.27重量%存在,并且
[0633][0634]
以小于0.01重量%存在。
[0635]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,在连续搅拌下将2.01份组合物(即在二甲基甲酰胺中25%固体时)添加到15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试19-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试19-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0636]
乙醇斑点测试
[0637][0638]
遗传毒性测试
[0639][0640]
实施例20
[0641]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(91.0克)、2-乙基己基缩水甘油醚(201.0克)和k2co3(10.00克)并加热至80℃,在此之后将混合物在t=80℃下搅拌47小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0642]
将130克所得物质与0.02克新癸酸铋和668克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将107.4克desmodur n 3600在668克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将10克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至75℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得高粘度无色液体。理论主组分的计
算分子量为1233.93da,化学结构如下所示。
[0643][0644]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1256.93da;观察到的[m+na+]=1256.86da。通过lc-ms测定并定量质量低于580da的以下组分:
[0645][0646]
在组合物中以0.84重量%存在,并且
[0647][0648]
以0.16重量%存在。
[0649]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.96份组合物与0.96份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试20-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试20-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0650]
乙醇斑点测试
[0651][0652][0653]
遗传毒性测试
[0654][0655]
实施例21
[0656]
向20ml反应小瓶中装入丙烯亚胺(1.98克)、邻甲苯缩水甘油醚(5.57克),加盖,加热至55℃,在此之后将混合物在t=55℃下搅拌20小时。真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0657]
将0.90克desmodur n 3600、0.02克新癸酸铋和5.80克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在5分钟内将0.99克来自第一步骤的产物在5.80克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将5.80克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空蒸发溶剂至43%固体,得到低粘度的淡黄色液体。理论主组分的计算分子量为1167.69da,化学结构如下所示。
[0658][0659]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1190.69da;观察到的[m+na+]=1190.77da。通过lc-ms测定并定量质量低于580da的以下组分:
[0660][0661]
在组合物中以0.08重量%存在,并且
[0662][0663]
以0.08重量%存在。
[0664]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,在连续搅拌下将0.47份组合物(即在二甲基甲酰胺中
43%固体时)添加到5份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试21-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试21-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0665]
乙醇斑点测试
[0666][0667]
遗传毒性测试
[0668][0669][0670]
实施例22
[0671]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0672]
将11.09克所得物质(新癸酸2-羟基-3-(2-甲基吖丙啶-1-基)丙基酯)与0.02克新癸酸铋和46克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将5.00克1,3-双(1-异氰酸根合-1-甲基乙基)苯在46克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的粘性液体。理论主组分的计算分子量为814.58da,化学结构如下所示。
[0673]
[0674]
通过maldi-tof-ms确认分子量:计算的[m+na+]=837.58da;观察到的[m+na+]=837.48da。通过lc-ms测定并定量质量低于580da的以下组分:
[0675][0676]
在组合物中以1.3重量%存在。
[0677]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.61份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.81份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试22-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试22-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0678]
乙醇斑点测试
[0679][0680]
遗传毒性测试
[0681][0682]
实施例23
[0683]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0684]
将10.33克所得物质(新癸酸2-羟基-3-(2-甲基吖丙啶-1-基)丙基酯)与0.02克新癸酸铋和43克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将5.00克4,4'-亚甲基双(环己基异氰酸酯)在43克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的粘性液体。理论主组分的计算分子量为832.63da,化学结构如下所示。
[0685][0686]
通过maldi-tof-ms确认分子量:计算的[m+na+]=855.62da;观察到的[m+na+]=855.52da。通过lc-ms测定并定量质量低于580da的以下组分:
[0687][0688]
在组合物中以0.2重量%存在。
[0689]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.64份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.83份所得溶液添加至21份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试23-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试23-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0690]
乙醇斑点测试
[0691][0692]
遗传毒性测试
[0693][0694]
实施例24
[0695]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0696]
将2.20克所得物质(新癸酸2-羟基-3-(2-甲基吖丙啶-1-基)丙基酯)与0.02克新癸酸铋和18克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在15分钟内将1.50克desmodur n 3600在18克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至70℃。以规律间
隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的高粘度液体。理论主组分的计算分子量为1359.96da,化学结构如下所示。
[0697][0698]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1382.95da;观察到的[m+na+]=1382.86da。通过lc-ms测定并定量质量低于580da的以下组分:
[0699][0700]
在组合物中以0.27重量%存在。
[0701]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.68份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.86份所得溶液添加至21份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试24-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试24-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0702]
乙醇斑点测试
[0703][0704]
遗传毒性测试
[0705]
[0706]
实施例25
[0707]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0708]
将配备有温度计和顶置式搅拌器的500ml圆底烧瓶置于n2气氛下,并装入desmodur w(60.08克)和65.35克先前步骤的产物。将所得混合物加热至50℃,在此之后添加新癸酸铋(0.05克)。使混合物放热,然后进一步加热至80℃并在80℃下搅拌2.5小时。然后向混合物中添加pthf650(74.52克),并将该混合物在80℃下再搅拌1小时。真空去除溶剂,以获得无色固体。
[0709]
理论主组分的计算分子量为832.63da(结构25-a:没有pthf650重复单元)、1473.1.0da(结构25-b:一个具有5个四亚甲基醚二醇重复单元的pthf区段)、1545.15da(结构25-c:一个具有6个四亚甲基醚二醇重复单元的pthf区段)和2257.68da(结构25-d:两个具有6个四亚甲基醚二醇重复单元的pthf区段),化学结构如下所示。
[0710]
结构25-a:
[0711][0712]
通过maldi-tof-ms确认分子量:结构25-a:计算的[m+na+]=855.63da;观察到的[m+na+]=855.66da。
[0713]
结构25-b:
[0714][0715]
通过maldi-tof-ms确认分子量:结构25-b:计算的[m+na+]=1496.10da;观察到的[m+na+]=1496.16da。
[0716]
结构25-c:
[0717][0718]
通过maldi-tof-ms确认分子量:结构25-c:计算的[m+na+]=1568.15da;观察到的[m+na+]=1568.21da。
[0719]
结构25-d:
[0720][0721]
通过maldi-tof-ms确认分子量:结构25-d:计算的[m+na+]=2280.68da;观察到的[m+na+]=2280.78da。
[0722]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将1.36份组合物与1.36份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试25-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试25-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0723]
乙醇斑点测试
[0724][0725]
遗传毒性测试
[0726][0727]
实施例26
[0728]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0729]
将配备有温度计和顶置式搅拌器的500ml圆底烧瓶置于n2气氛下,并装入desmodur w(49.60克)和53.95克先前步骤的产物。将所得混合物加热至50℃,在此之后添加新癸酸铋(0.04克)。使混合物放热,然后进一步加热至80℃并在80℃下搅拌2小时。然后向该混合物中添加durez-ter s 105-110(96.40克)。在80℃下再搅拌2.5小时后,向该混合物中添加第二批durez-ter s 105-110(10.00克),并将该混合物在80℃下再搅拌11小时。真空去除溶剂,以获得白色固体。
[0730]
理论主组分的计算分子量为832.63da(结构26-a)、1212.90(结构26-b:来自durez-ter s 105-110的两个desmodur w基团之间的一个己二醇连接基团)、1441.03da(结构26-c:1个聚酯重复单元)、1669.17da(结构26-d:2个聚酯重复单元)、1897.30da(结构26-e:3个聚酯重复单元)和2125.44da(结构26-f:4个聚酯重复单元),化学结构如下所示。
[0731]
结构26-a:
[0732][0733]
通过maldi-tof-ms确认分子量:结构26-a:计算的[m+na+]=855.63da;观察到的[m+na+]=855.70da。
[0734]
结构26-b:
[0735][0736]
通过maldi-tof-ms确认分子量:结构26-b:计算的[m+na+]=1235.90da;观察到的[m+na+]=1236.01da。
[0737]
结构26-c:
[0738][0739]
通过maldi-tof-ms确认分子量:结构26-c:计算的[m+na+]=1464.03da;观察到的[m+na+]=1464.16da。
[0740]
结构26-d:
[0741][0742]
通过maldi-tof-ms确认分子量:结构26-d:计算的[m+na+]=1692.17da;观察到的[m+na+]=1692.32da。
[0743]
结构26-e:
[0744][0745]
通过maldi-tof-ms确认分子量:结构26-e:计算的[m+na+]=1920.30da;观察到的[m+na+]=1920.48da。
[0746]
结构26-f:
[0747][0748]
通过maldi-tof-ms确认分子量:结构26-f:计算的[m+na+]=2148.44da;观察到的[m+na+]=2148.62da。
[0749]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将1.73份组合物与1.73份乙酸1-甲氧基-2-丙基酯混合,并在定期搅拌下在80℃下孵育10分钟。随后,在连续搅拌下将所得溶液添加至15份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试26-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试26-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0750]
乙醇斑点测试
[0751][0752][0753]
遗传毒性测试
[0754][0755]
实施例27
[0756]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0757]
将2.89克所得物质(新癸酸2-羟基-3-(2-甲基吖丙啶-1-基)丙基酯)与0.02克新癸酸铋、1.35克平均mn为500da的聚(乙二醇)单甲醚和30克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将2.5克desmodur n 3600在30克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得澄清的高粘度液体。理论主组分的计算分子量为1359.96da(三个氮丙啶)和1591.04da(两个氮丙啶,11eg重复单元),化学结构如下所示。
[0758][0759]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.93份的组合物与0.60份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将1.02份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试27-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试27-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0760]
乙醇斑点测试
[0761][0762][0763]
遗传毒性测试
[0764][0765]
实施例28
[0766]
将配备有冷凝器的1l圆底烧瓶置于n2气氛下,并装入丙烯亚胺(69.0克)、cardura e10p(201.0克)和k2co3(7.30克)并加热至80℃,在此之后将混合物在t=80℃下搅拌24小时。在过滤后,真空去除过量的pi,得到无色的低粘度液体。
[0767]
将19.49克所得物质(新癸酸2-羟基-3-(2-甲基吖丙啶-1-基)丙基酯)与0.02克新癸酸铋、37.94克平均mn为2000da的聚(乙二醇)单甲醚和11.07克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在45分钟内将17.55克desmodur n 3600在11.07克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,再将10克2-甲基四氢呋喃通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得不透明的高粘度液体。理论主组分的计算分子量为1359.96da(三个氮丙啶)、3043.91da(两个氮丙啶,44eg重复单元)、3087.94da(两个氮丙啶,45eg重复单元)和3131.96da(两个氮丙啶,46eg重复单元),化学结构如下所示。
[0768][0769]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1382.96da;观察到的[m+na+]=1382.91da。
[0770][0771]
通过maldi-tof-ms确认分子量:计算的[m+na+]=3066.91da;观察到的[m+na+]=3066.77da。
[0772][0773]
通过maldi-tof-ms确认分子量:计算的[m+na+]=3110.94da;观察到的[m+na+]=3110.79da。
[0774][0775]
通过maldi-tof-ms确认分子量:计算的[m+na+]=3154.96da;观察到的[m+na+]=3154.81da。通过lc-ms测定并定量质量低于580da的以下组分:
[0776][0777]
在组合物中以0.02重量%存在。
[0778]
遗传毒性测试
[0779][0780]
实施例29
[0781]
将配备有冷凝器的100ml反应器置于n2气氛下,并装入丙烯亚胺(2.40克)、mw为550da的mpeg-环氧化物(10.1克)、2-甲基四氢呋喃(20ml)和k2co3(1.1g),并加热至80℃,然后将该混合物在t=80℃下搅拌25小时。在过滤后,真空去除溶剂和过量的pi,得到深棕色粘性油状物。
[0782]
将3.57克所得物质(mw为550da的1-(ω-甲氧基(低聚环氧乙烷))-3-(2-甲基吖丙啶-1-基)丙-2-醇)与0.02克新癸酸铋和21克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在15分钟内将1.00克desmodur n 3600在21克二甲基甲酰胺中的溶液滴加到反应烧瓶中,在此之后将混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得褐色的高粘度液体。理论主组分的计算分子量为2656.62da,化学结构如下所示。
[0783][0784]
基于来自din 68861-1标准的工序使用涂层表面上的斑点测试来评定合成的化合物作为交联剂的性能。对于这些测试,将0.70份的组合物与0.39份proglyde
tm
dmm(二丙二醇二甲醚,异构体的混合物)混合,并在80℃下在定期搅拌下孵育10分钟。随后,在连续搅拌下将0.73份所得溶液添加至20份r-1005中,并将所得混合物进一步搅拌30分钟。然后,将该涂料组合物过滤,并使用100μm线材涂布器施加到leneta测试卡上(测试29-1)。作为参考,还从缺乏交联剂的相同组合物流延成膜(测试29-2)。将膜在25℃下干燥16小时,然后在50℃下退火1小时,并在25℃下进一步干燥24小时。随后,将一块脱脂棉浸入1:1etoh:脱矿质水中,并在膜上放置各种时间间隔。在去除etoh并恢复60分钟后,获得了以下结果(评分1指示膜的完全降解,10指示没有可见的损伤):
[0785]
乙醇斑点测试
[0786][0787]
遗传毒性测试
[0788][0789]
实施例30
[0790]
将配备有冷凝器的100ml反应器置于n2气氛下,并装入丙烯亚胺(1.30克)、mw为1kda的mpeg-环氧化物(10.1克)、2-甲基四氢呋喃(50ml)和k2co3(1.1g),并加热至80℃,然后将该混合物在t=80℃下搅拌25小时。在过滤后,真空去除溶剂和过量的pi,得到浅棕色粘性液体。
[0791]
将5.61克所得物质(mw为1kda的1-(ω-甲氧基(低聚环氧乙烷))-3-(2-甲基吖丙啶-1-基)丙烷-2-醇)与0.02克新癸酸铋和13.79克2-甲基四氢呋喃一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在25分钟内将1.00克desmodur n 3600在13.79克2-甲基四氢呋喃中的溶液滴加到反应烧瓶中,再将10克2-甲基四氢呋喃通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至70℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸为止。真空去除溶剂,以获得褐色的高粘度液体。理论主组分的计算分子量为4241.57da,化学结构如下所示。
[0792][0793]
遗传毒性测试
[0794][0795]
实施例31
[0796]
将配备有冷凝器的100ml反应器置于n2气氛下,并装入2,2-二甲基氮丙啶(8.00克)、正丁基缩水甘油醚(10.0克)、2-甲基四氢呋喃(20ml)和k2co3(0.75克),并在30分钟内加热到80℃,然后将该混合物在t=80℃下搅拌44小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0797]
将0.26克所得物质(1-丁氧基-3-(2,2-二甲基吖丙啶-1-基)丙-2-醇)与0.01克新癸酸铋和3克二甲基甲酰胺一起装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在10分钟内将0.26克desmodur n 3600在3克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将1克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.03克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。该混合物为不透明的液体。理论主组分的计算分子量为1107.79da,化学结构如下所示。
[0798][0799]
通过maldi-tof-ms确认分子量:计算的[m+na+]=1130.79da;观察到的[m+na+]=1130.86da。通过lc-ms测定并定量质量低于580da的以下组分:
[0800][0801]
在组合物中以小于0.01重量%存在并且
[0802][0803]
在组合物中以0.89重量%存在。
[0804]
遗传毒性测试
[0805][0806]
实施例32
[0807]
向反应小瓶中装入2-乙基氮丙啶(2.50克)、正丁基缩水甘油醚(2.00克)和k2co3(0.5克),加盖并加热至80℃,然后将混合物在t=80℃下搅拌20小时。在过滤后,真空去除过量的pi,之后通过真空蒸馏进一步纯化,得到无色的低粘度液体。
[0808]
将4.0克desmodur n 3600、0.02克新癸酸铋和8.10克二甲基甲酰胺装入配备有温度计的反应烧瓶中。将混合物在氮气氛下用机械上部搅拌器搅拌并加热至50℃。然后在10分钟内将4.0克来自第一步骤的产物在8.10克二甲基甲酰胺中的溶液滴加到反应烧瓶中,再将8.10克二甲基甲酰胺通过进料漏斗冲入该反应混合物中,然后将该混合物进一步加热至80℃。以规律间隔取样,并使用bruker alpha ft-ir光谱仪监测反应进程,直到在2200-2300cm-1
处观察不到nco拉伸变化为止。随后,将0.13克的1-丁醇添加至混合物中,随后进一步反应以使上述nco拉伸峰完全消失。该混合物为澄清液体。理论主组分的计算分子量为1107.79da,化学结构如下所示。
[0809][0810]
遗传毒性测试
[0811]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1