基因治疗DNA载体GDTT1.8NAS12及用于获得它的方法与流程
发明领域
本发明涉及基因工程并且可用于生物技术、医学和农业中制作基因治疗产品。也就是说,可以利用生产的含有治疗性基因的基因治疗dna载体将其递送至经历该基因表达减少或不足的人和动物的细胞中,从而确保所期望的治疗效果。
发明背景
基因治疗涉及引入核酸或其衍生物来治疗后天或遗传性疾病。治疗效果是通过补偿功能异常或完全无功能的基因,或通过生产具有治疗效果的蛋白质产物来实现的。基因治疗的主要工具是治疗性遗传物质载体(carrier),即载体(vector)。所有用于基因治疗的载体分为两大类:病毒性和非病毒性载体。病毒性基因治疗载体基于逆转录病毒、慢病毒、腺病毒、腺相关病毒、疱疹病毒和痘病毒构建。(lundstromk.viralvectorsingenetherapy.diseases.2018may21;6(2))。非病毒性载体包括基于质粒或线性rna的载体。质粒载体因其制作简化且稳定性高,在实践中应用较为广泛。为了提高治疗性基因递送到体细胞的效率,常引入质粒载体与不同的载体相组合,所述载体如脂质、阳离子聚合物、树枝状聚合物、多肽和不同性质的纳米颗粒等(hidaic,kitanoh.nonviralgenetherapyforcancer:areview.diseases.2018jul3;6(3))。添加至此类分子复合物的质粒可以提高其对细胞的渗透力。
病毒载体的主要优势是由于病毒的天然特性,快速且高效地将治疗性遗传物质递送至细胞中。但是,在临床实践中使用的病毒载体有很大的局限性。除了与获得、生产和选择有关的纯技术问题外,病毒载体的应用还涉及高风险的不良医疗后果,如炎症和对基因治疗的免疫应答、细胞毒性、突变发生和致癌作用。病毒载体的另一个风险是患者的具体个体特征可能导致基因治疗的不可预知的后果(thomasce,ehrhardta,kayma.progressandproblemswiththeuseofviralvectorsforgenetherapy.natrevgenet.2003may;4(5):346-58)。尽管密集的研究旨在降低使用病毒载体进行基因治疗的相关风险,但许多问题仍未解决。在这方面,研究人员和临床医生对非病毒载体进行治疗性基因递送的应用越来越感兴趣。
质粒是一种独立于染色体dna而存在并在细胞中复制的环状dna分子。在体内,质粒主要存在于细菌中,且较少存在于古细菌和真核生物中。染色体dna携带着在正常条件下生命所需的全部遗传信息,质粒通常包含确保在特定或甚至不利环境中存活的基因(lippsg.(editor),(2008).plasmids:currentresearchandfuturetrends.caisteracademicpress.isbn978-1-904455-35-6)。此类基因确保抗生素抗性,编码毒力因子,参与各种底物的分解代谢和新陈代谢以及有害化合物的解毒。已知质粒参与了细胞之间的遗传信息转移,从而实现水平基因转移。
基于质粒的载体不共享病毒载体的缺点。因此,由于直接分子克隆和产生所需量的简单方法,质粒就其性质而言是用于治疗性遗传物质的便捷载体。质粒载体引入不会引起受试者的炎症和免疫应答。质粒本身不具有细胞毒性,并且当其到达靶细胞时不会整合到基因组中,因此其不会影响基因组稳定性。由于这些特点,质粒载体是用于基因治疗和遗传预防治疗(dna疫苗)的非常有前景的工具(porterkr,raviprakashk.dnavaccinedeliveryandimprovedimmunogenicity.currissuesmolbiol.2017;22:129-138)。
此外,已经积累了相当多的用质粒载体操作的经验,因为数十年来,质粒载体一直是科学和生物技术实验室中分子克隆和重组蛋白获得的主要工具(russel,davidw.;sambrook,joseph(2001),molecularcloning:alaboratorymanual.coldspringharbor,n.y;coldspringharborlaboratory.)。
然而,质粒载体也具有一些缺点,限制了其在临床实践中的应用。首先,要将抗生素抗性基因引入质粒载体以用于在载体菌株中进行选择和生产。其次,将调控元件(启动子、增强子、转录后调控元件)添加至载体以用于治疗性基因的有效表达(sunj,lid,haoy,zhangy,fanw,fuj,huy,liuy,shaoy.posttranscriptionalregulatoryelementsenhanceantigenexpressionanddnavaccineefficiency.dnacellbiol.2009may;28(5):233-40),其主要构成病毒核苷酸序列(draftguidelineonthequality,non-clinicalandclinicalaspectsofgenetherapymedicinalproducts,http://www.ema.europa.eu/docs/en_gb/document_library/scientific_guideline/2015/05/wc500187020.pdf)。最后,质粒载体的大小决定了渗透到靶细胞中的效率,即载体越大,其渗透到细胞越差。基因治疗中使用的质粒载体通常具有不必要的非编码位点,大大增加了其长度(mairhoferj,grabherrr.rationalvectordesignforefficientnon-viralgenedelivery:challengesfacingtheuseofplasmiddna.molbiotechnol.2008.39(2):97-104)。
近年来,感染原的抗生素抗性的增加是抗微生物药物在医学中广泛使用的自然反应。对抗生素抗性的增加具有重大的社会和经济意义,并被视为对国家安全的威胁(macphersond.w.,gushulakb.d.,bainew.b.,balas.,gubbinsp.o.,holtomp.,segarra-newnhamm.2009.populationmobility,globalization,andantimicrobialdrugresistance.emerginfectdis15:1727-1732)。在这方面,使用添加抗生素抗性基因的质粒载体进行选择和生产有将此类基因直接或通过水平转移转移到感染原的风险(sanmillana.evolutionofplasmid-mediatedantibioticresistanceintheclinicalcontext.trendsmicrobiol.2018jul23.doi:10.1016/j.tim.2018.06.007)。因此,欧洲药品管理局建议不要将抗生素抗性基因加入质粒基因治疗载体(关于开发过程中基因治疗医药产品设计修改的反思文件/2011年12月14日ema/cat/gtwp/44236/2009先进疗法委员会)。
将必要的抗生素抗性基因引入质粒载体的问题可以通过开发的大肠杆菌菌株(dh1lacdapd和dh1lacp2dapd)来避免,所述大肠杆菌菌株的特征在于其基因组中的dapd基因受lac启动子控制(cranenburghrm,hanakja,williamssg,sherrattdj.escherichiacolistrainsthatallowantibiotic-freeplasmidselectionandmaintenancebyrepressortitration.nucleicacidsres.2001.29(5):e26)。该基因编码2,3,4,5-四氢吡啶-2,6-二羧酸-n-琥珀酰转移酶,该酶参与l-赖氨酸的生物合成。在没有激活dapd表达的诱导物iptg(异丙基-β-d-1-硫代吡喃半乳糖苷)情况下,这些菌株经受裂解。dapd的表达也可以通过引入多拷贝port载体来诱导,使得可以选择和繁殖转化的克隆。这些菌株的缺点是转化水平低和转化不稳定。
避免使用抗生素抗性基因的另一种方法涉及修饰大肠杆菌细胞,使得rnai质粒载体复制抑制剂可以通过形成rna/反义rna双链体抑制对细菌生长至关重要的基因的翻译(如mura,其编码udp-n-乙酰氨基葡萄糖1-羧基乙烯基转移酶,其参与细菌细胞壁肽聚糖的生物合成)(mairhoferj,pfaffenzelleri,merzd,grabherrr.anovelantibioticfreeplasmidselectionsystem:advancesinsafeandefficientdnatherapy.biotechnolj.2008.3(1):83-89)。mura基因应受tetr阻遏蛋白的控制,并且只能在构建的含有rna-i基因的质粒载体存在下表达,然而,在该系统中,抑制的机制仍然是未知的,并且iptg介导的诱导导致缺乏治疗性质粒载体的大肠杆菌菌落的形成。
已经描述了避免在质粒基因治疗载体的构建中应用抗生素抗性基因的某些其他方式(mignonc,sodoyerr,werleb.antibiotic-freeselectioninbiotherapeutics:nowandforever.pathogens.2015apr3;4(2):157-81)。
为了创造不含非功能序列的最小载体,已获得缺乏任何原核生物核苷酸序列的超螺旋质粒dna分子。这种所谓的微形环状载体只含有一个复制起点和抗生素抗性基因,并且通过使用
专利权利要求us2011152377/10提出使用缺乏抗生素抗性基因但仍编码阻遏蛋白的质粒载体。这种阻遏蛋白抑制大肠杆菌的染色体dna中编码的毒性蛋白的表达。然而,此类转化的低效率及不稳定限制了在工业规模上具有阻遏蛋白的载体的生产。
专利us7521182b2提出含有arad基因的质粒载体。在大肠杆菌中,该基因编码l-核糖-5-磷酸-4-表异构酶。虽然这种酶本身对细菌生长并不重要,但它的缺乏会导致有毒产物在细胞中的积累。编码arad的载体与从基因组中去除该基因的大肠杆菌菌株组合代表了无抗生素的选择系统。
专利us9,644,211提出了一种获得不含原核生物序列的微形环状质粒载体的方法。此类载体是由在培养的大肠杆菌菌株中实现的para介导的重组产生的。由于该微形环不可能在工业规模上产生,因此无法将其视为潜在的基因治疗载体。
基于重组质粒基因治疗载体的新载体的原型是用于基因接种的重组载体(us9,550,998)。新的载体是超螺旋质粒,其用于在人和动物细胞中表达克隆的基因。它由复制起点、人巨细胞病毒启动子和增强子以及来自人t嗜淋巴细胞病毒的调控元件组成。
通过反义互补由噬菌体引入菌株的sacb基因,选择并在特殊用途的大肠杆菌菌株中生产新载体,从而完全排除了抗生素抗性基因。限制新载体用于基因治疗的唯一因素是在包括病毒核苷酸序列形式的调控元件。
发明公开
本发明的目的是构建一种用于人和动物细胞遗传修饰的多功能基因治疗dna载体,其将合理地组合以下各项:
i)由于不超过2600bp的有限长度,基因治疗dna载体增强各种人和动物组织细胞中治疗性基因表达水平的效率,从而确保有效渗透到靶细胞中以及确保人和动物组织细胞中的高表达的调控元件序列的可用性。
ii)由于基因治疗dna载体中不存在代表病毒基因组的核苷酸序列的调控元件,安全应用于人类和动物的基因治疗的可能性。
iii)由于基因治疗dna载体中不存在抗生素抗性基因,安全应用于人类和动物的基因治疗的可能性。
iv)在工业规模上基因治疗dna载体的可生产性和可构建性。
根据国家监管机构对基因治疗药物的建议,特别是欧洲药品管理局拒绝将抗生素抗性标记加入新工程化改造的用于基因治疗的质粒载体的要求(关于基因治疗医药产品在开发过程中设计修改的反思文件/2011年12月14日ema/cat/gtwp/44236/2009先进疗法委员会),以及拒绝将病毒基因组加入新工程化改造的用于基因治疗的质粒载体的要求(基因治疗医药产品的质量、非临床和临床方面的准则/2015年3月23日,ema/cat/80183/2014,先进疗法委员会)在本文中提供项ii和iii。
本发明的目的还包括构建携带该基因治疗dna载体的菌株,以便在工业规模上生产这些基因治疗dna载体。
通过创建形式为含有核苷酸序列seqidno.1的2591-bp环状双链dna分子的基因治疗dna载体gdtt1.8nas12可以达到指定的目的,所述基因治疗dna载体能够在大肠杆菌细胞中自主复制,并由以下结构元件组成:在基因的第一内含子中包含的ef1a人延伸因子的启动子区与其自己的增强子,含有bamhi、ecorv、sali、kpni、ecori、xbai和noti限制性位点的序列且设计用于克隆治疗性治疗基因的多接头,转座子tn10的rna-out调控元件,其使得能够对大肠杆菌菌株jm110-nas进行无抗生素阳性选择;复制起点,其用于基因治疗dna载体的自主复制,所述复制起点具有单个核苷酸取代以增加大多数大肠杆菌菌株的细胞中的质粒产量。
获得2591-bp基因治疗dna载体gdtt1.8nas12的方法涉及首先构建包含688-bp复制起点,467-bp的转录终止子hgh-ta,137-bp的转座子tn10的调控位点rna-out,1018-bp的卡那霉素抗性基因和68-bp的多接头的2408-bp的中间载体,进一步使用sali和bamhi限制性内切核酸酶进行载体分割,并与含有人延伸因子ef1a的启动子区与其自己的1219-bp的增强子的启动子/调控位点连接,以及通过spei限制性位点切割所述卡那霉素抗性基因。
获得大肠杆菌菌株jm110-nas以用于生产基因治疗dna载体gdtt1.8nas12的方法涉及构建线性dna片段,其含有允许无抗生素阳性选择的tn10转座子的64-bp调控元件rna-in,1422-bp的sacb果聚糖蔗糖酶基因,其产物确保在含蔗糖的培养基内的选择,对挑取其中发生同源重组的菌株克隆所需的763-bp的catr氯霉素抗性基因,以及确保reca基因的区域中的同源重组,同时基因失活的两个同源序列(329bp和233bp),其中所述同源序列通过reca基因片段的pcr扩增获得,所述pcr扩增使用大肠杆菌jm110-nas的基因组dna为基质以及引物对lha-f(5’-gctgacgctgcaggtgatc)和lha-r(5’-gacaagatgtgtgtctaccgcttcaggttacccgccag),及引物对rha-f(5’-tggcagggcggggcgtaactacgcctctgttcgtctcga)和rha-r(5’-ctcagcagcaactcacgtac)进行,之后通过电穿孔转化大肠杆菌细胞,并选择在含有10μg/ml氯霉素的培养基中存活的克隆。
通过上述方法获得的大肠杆菌菌株jm110-nas,用于在可能无抗生素阳性选择的情况下生产基因治疗dna载体gdtt1.8nas12,所述大肠杆菌菌株jm110-nas在染色体中在reca基因区中含有线性片段,所述线性片段由转座子tn10的调控元件rna-in、sacb果聚糖蔗糖酶基因和catr氯霉素抗性基因组成。
获得携带基因治疗dna载体gdtt1.8nas12的大肠杆菌菌株jm110-nas/gdtt1.8nas12的方法,其涉及制备大肠杆菌菌株jm110-nas的电感受态细胞,并对这些细胞进行基因治疗dna载体gdtt1.8nas12的电穿孔。之后,将所述细胞倒入具有选择性培养基的琼脂平板(培养皿)上,所述选择性培养基含有酵母浸膏、蛋白胨、6%蔗糖和10μg/ml氯霉素。
通过上述方法获得的大肠杆菌菌株jm110-nas/gdtt1.8nas12,其携带基因治疗dna载体gdtt1.8nas12用于生产允许无抗生素选择的基因治疗dna载体gdtt1.8nas12。
在工业规模上基因治疗dna载体gdtt1.8nas12生产的方法涉及在工业发酵罐中将大肠杆菌菌株jm110-nas/gdtt1.8nas12的细菌培养物放大至增加细菌生物质所需的量,其后使用所述生物质提取出级份,所述级份含有治疗dna产物,即基因治疗dna载体gdtt1.8nas12,然后进行多级过滤并通过层析方法进行纯化。
在附图中解释了本发明的实质,其中
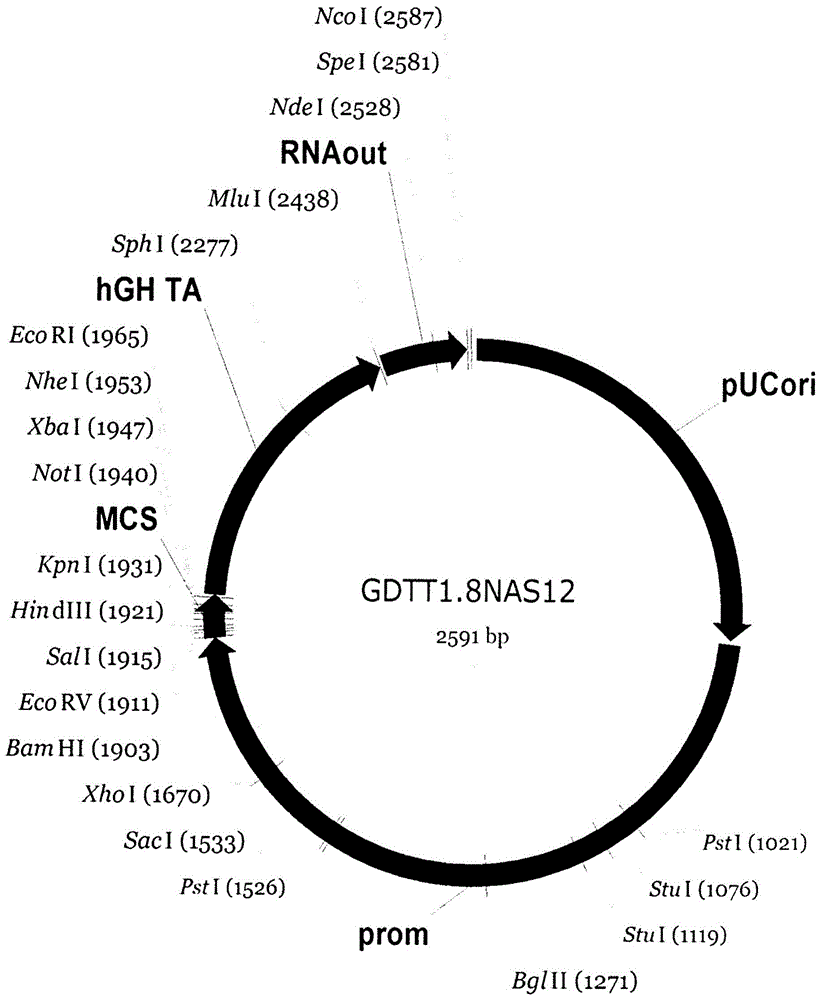
图1
显示基因治疗dna载体gdtt1.8nas12的结构,其是2591-bp的环状双链dna分子,其能够在大肠杆菌细胞中自主复制。
图1中标出了载体的以下结构元件:
(1)prom(695至1901bp)是基因的第一内含子中包含的人延伸因子ef1a的启动子区与内在的增强子。其确保大多数人组织中重组基因的有效转录。
(2)mcs(1902至1969bp)是多接头(多克隆位点),其含有bamhi、ecorv、sali、kpni、ecori、xbai和noti限制性酶的序列,并允许治疗性治疗基因的克隆。
(3)rna-out(2443至2579bpout)是转座子tn10的调控元件rna-out,其在利用大肠杆菌菌株jm110的情况下允许无抗生素阳性选择。
(4)ori(1至688bp)是用于自主复制的复制起点,其具有单个核苷酸取代以增加大多数大肠杆菌菌株的细胞中的质粒产量。
图2
显示大肠杆菌的基因reca区中同源重组以获得大肠杆菌菌株jm110的dna片段的结构。
线性片段由盒组成,所述盒携带用于无抗生素选择的转座子tn10的调控元件rna-in(64bp),sacb果聚糖蔗糖酶基因,其产物确保在含蔗糖的培养基内进行选择(1422bp),以及对挑取其中发生同源重组的菌株克隆所需的catr(763bp)。盒侧翼为两个同源臂,其确保reca基因区域中的重组的过程同时基因失活(左臂和右臂分别为329bp和233bp)。
图3
显示用pefgp-c1质粒载体(clontech)和dna载体gdtt1.8nas12-egfp转染细胞后48小时mg-63细胞培养物的荧光显微成像(a),和用pefgp-c1质粒载体(clontech)和dna载体gdtt1.8nas12-egfp转染细胞后48小时从mg-63细胞中提取的蛋白质发出的荧光图(b),以比较用pefgp-c1质粒载体(clontech)和dna载体gdtt1.8nas12-egfp转染细胞后48小时mg-63细胞中靶基因产物(例如绿色荧光蛋白(gfp))的积累水平。
图4
显示用携带人谷胱甘肽过氧化物酶1基因的dna载体gdtt1.8nas12-gpx1转染前和转染细胞后48小时,人谷胱甘肽过氧化物酶1基因mrna在表皮角质形成细胞heka的原代培养细胞中的积累的图,以评估用携带人谷胱甘肽过氧化物酶1基因的dna载体gdtt1.8nas12-gpx1转染前和转染细胞后48小时,heka表皮角质形成细胞的原代培养细胞中例如谷胱甘肽过氧化物酶1基因的治疗性基因mrna积累,其中:
1-用基因治疗载体gdtt1.8nas12转染后gpx1基因的cdna,
2-用携带人谷胱甘肽过氧化物酶1基因的区域的基因治疗载体gdtt1.8nas12-gpx1转染后gpx1基因的cdna,
3-用基因治疗载体gdtt1.8nas12转染后b2m基因的cdna,
4-用携带人谷胱甘肽过氧化物酶1基因的区域的基因治疗载体gdtt1.8nas12-gpx1转染后b2m基因的cdna。
图5
显示用携带人谷胱甘肽过氧化物酶1基因编码区的基因治疗载体gdtt1.8nas12-gpx1注射到3名患者的皮肤后,这些患者的皮肤活检样品中谷胱甘肽过氧化物酶1蛋白浓度的图,其目的在于分析用携带治疗性基因(例如人谷胱甘肽过氧化物酶1基因)的基因治疗载体gdtt1.8nas12-gpx1注射人皮肤后,人皮肤中谷胱甘肽过氧化物酶1蛋白浓度的变化,其中:
p1i是基因治疗dna载体gdtt1.8nas12-gpx1注射区域中的患者p1皮肤活检,
p1ii是基因治疗dna载体gdtt1.8nas12(安慰剂)注射区域中的患者p1皮肤活检。
p1iii是来自完整部位的患者p1皮肤活检。
p2i是基因治疗dna载体gdtt1.8nas12-gpx1注射区域中的患者p2皮肤活检,
p2ii是基因治疗dna载体gdtt1.8nas12(安慰剂)注射区域中的患者p2皮肤活检,
p2iii是来自完整部位的患者p2皮肤活检,
p3i是基因治疗dna载体gdtt1.8nas12-gpx1注射区域中的患者p3皮肤活检,
p3ii是基因治疗dna载体gdtt1.8nas12(安慰剂)注射区域中的患者p3皮肤活检,
p3iii是来自完整部位的患者p3皮肤活检。
图6
显示用携带编码绿色荧光蛋白的基因位点的dna载体gdtt1.8nas12和dna载体gdtt1.8nas12-egfp转染后48小时的benepc牛子宫内膜上皮细胞的原代培养物中绿色荧光蛋白的荧光水平变化的图,以比较靶基因产物(例如绿色荧光蛋白(gfp))在用携带编码绿色荧光蛋白的基因位点的dna载体gdtt1.8nas12和dna载体gdtt1.8nas12-egfp转染后48小时benepc牛子宫内膜上皮细胞的原代培养物中的积累水平。
本发明的实施方案
本发明体现如下。
首先,构建了2591-bp的基因治疗dna载体gdtt1.8nas12,用于对动物和人细胞进行遗传修饰,其含有核苷酸序列seqidno.1。获得2591-bp的基因治疗dna载体gdtt1.8nas12的方法涉及,首先,构建一个2408-bp的载体,其包含688-bp的复制起点、467-bp的转录终止子hgh-ta、137-bp的转座子tn10的调控位点rna-out、1018-bp的卡那霉素抗性基因和68-bp的多接头,进一步地使用sali和bamhi限制性内切核酸酶将载体分割,并连接到含有ef1a人延伸因子基因的启动子区域与其自己的1219-bp的增强子的启动子/调控位点,并通过spei限制性位点将卡那霉素抗性基因切割。
同时,本领域的技术人员知道遗传密码的简并性,其意指本发明的范围还包括核苷酸序列的变体,其差异在于核苷酸的插入、缺失或替换,但不导致改变由治疗性基因编码的多肽序列和/或不导致gdtt1.8nas12载体和/或基于它的携带治疗性基因的基因治疗dna载体的功能活性的丧失。本领域的技术人员意识到,获得基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗dna载体的方法论的实施可以在分子基因克隆的已知方法的选择内有所不同,并且这些方法包括在本发明的范围内。例如,可以使用不同的寡核苷酸序列、不同的限制性内切核酸酶或实验室技术,如不依赖连接的基因克隆,来扩增基因。
为了生产基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗dna载体(具有可能的无抗生素阳性选择),构建了大肠杆菌菌株jm110-nas。获得生产基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗dna载体的大肠杆菌菌株jm110-nas的方法涉及构建线性dna片段,其含有用于无抗生素阳性选择的64-bp的tn10转座子的调控元件rna-in,1422-bp的果聚糖蔗糖酶基因sacb,其产物确保在含蔗糖的培养基中的选择,对选择其中发生同源重组的菌株克隆所需的763-bp的氯霉素抗性基因catr,以及确保基因reca的区域中的同源重组过程,同时基因失活的两个同源序列(329bp和233bp),并进一步通过电穿孔转化大肠杆菌jm110细胞,并选择在含有10μg/ml氯霉素的培养基中存活的克隆。该方法还涉及构建携带基因治疗dna载体gdtt1.8nas12和/或基于它的含有治疗性基因的基因治疗dna载体的大肠杆菌菌株jm110-nas/gdtt1.8nas12,以用于生产允许无抗生素选择的载体。获得携带基因治疗dna载体gdtt1.8nas12和/或基于它的包含治疗性基因的基因治疗dna载体的大肠杆菌菌株jm110-nas/gdtt1.8nas12的方法涉及获得大肠杆菌菌株jm110-nas的电感受态细胞并用基因治疗dna载体gdtt1.8nas12对其进行电穿孔,进一步接种于具有选择性琼脂培养基的培养皿中,所述选择性琼脂培养基含有酵母浸膏、蛋白胨、6%蔗糖和10μg/ml氯霉素。
在工业规模上进行基因治疗dna载体gdtt1.8nas12生产的方法涉及在工业发酵罐中将菌株的细菌培养物放大至增加细菌生物质所需的量,其后使用生物质提取出级份,所述级份含有治疗性dna产物,即基因治疗dna载体gdtt1.8nas12,然后进行多级过滤并通过层析方法进行纯化。
本领域的技术人员认为,生产者菌株的培养条件、营养培养基的组成(除无抗生素外)、使用的设备以及dna纯化方法可以取决于特定生产线在标准操作程序的框架内有所不同,但已知的使用大肠杆菌菌株jm110-nas/gdtt1.8nas12进行dna载体的规模化、工业化生产和纯化的方法落入本发明的范围内。
本发明的实质通过以下实施用例进行说明。
实施例1.
获得基因治疗dna载体gdtt1.8nas12,该载体含有人延伸因子基因ef1a的启动子与其自己的增强子,以改善大多数人和动物细胞的细胞中治疗性基因的表达。
通过合并6个衍生自不同来源的dna片段构建基因治疗dna载体gdtt1.8nas12:
(a)复制起点,通过使用ucori-bam和ucori-nco寡核苷酸(序列表,(1)-(2))对市售puc19质粒的区域进行pcr扩增产生,
(b)hgh-ta转录终止子,通过使用hgh-f和hgh-r寡核苷酸(序列表,(3)和(4))对人基因组dna的位点进行pcr扩增产生,
(c)转座子tn10的调控位点rna-out,从ro-f、ro-r、ro-1、ro-2、ro-3寡核苷酸(序列表,(5)-(9))合成,
(d)卡那霉素抗性基因,通过使用kan-f和kan-r寡核苷酸(序列表,(10)和(11))对市售pet-28质粒的位点进行pcr扩增产生,
(e)多接头,通过对四种合成寡核苷酸mcs1、mcs2、mcs3和mcs4(序列表,(12)-(15))进行cining和退火产生,
(f)人延伸因子基因ef1a的启动子/调控位点与自身的增强子,通过利用ef1-xho和ef1-r寡核苷酸(序列表,(16)-(17))对人基因组dna位点进行pcr扩增产生。
根据制造商的说明,使用市售的试剂盒
实施例2.
为了证明dna载体gdtt1.8nas12的有效性,将治疗性基因,例如绿色荧光蛋白(gfp)基因克隆到多接头。
获得携带靶基因,例如编码绿色荧光蛋白(gfp)的基因,的基因治疗dna载体gdtt1.8nas12-egfp。
通过使用mvgfp-f和mvgfp-r寡核苷酸(序列表,(18)和(19))对市售质粒pegfp-c1(clontech)进行pcr扩增来产生绿色荧光蛋白基因的编码区。将获得的pcr片段通过bamhi和ecori限制性内切核酸酶切割,并与携带卡那霉素抗性基因的3608-bpdna载体连接,并用同样的酶切割。此外,如实施例1所述,从产生的载体中去除卡那霉素抗性基因。结果,得到了3268-bp的dna载体gdtt1.8nas12-egfp,其允许无抗生素的选择。
实施例3.
为了证明dna载体gdtt1.8nas12的有效性,将治疗性基因,例如人谷胱甘肽过氧化物酶1(gpx1)基因克隆到多接头。
获得携带治疗性基因的位点,如编码人谷胱甘肽过氧化物酶1蛋白的gpx1基因的dna载体gdtt1.8nas12-gpx1。
通过从患者的皮肤活检样品提取出总rna,然后进行逆转录反应和pcr扩增来产生gpx1基因的609-bp编码区(seqidno.2)。使用皮肤活检设备epitheasy3.5(medaxsrl)从前臂完整皮肤取样材料。用无菌生理溶液初步冲洗患者皮肤并用利多卡因溶液麻醉。活检样品的质量约为10mg。将样品置于1mltrizol试剂(thermofisherscientific)中,均质化,在65℃下加热5分钟。然后将样品在14,000g离心10分钟,并在65℃下再次加热10分钟。然后加入200μl氯仿,轻轻搅拌并在14,000g离心10分钟。然后分离水相,与1/10体积的3m醋酸钠ph5.2和等体积的异丙醇混合。将样品在-20℃下孵育10分钟,然后在14,000g下离心10分钟。沉淀的rna用1ml的70%的乙醇冲洗,在空气中干燥,并溶解在10μl不含rnase的水中。为了合成人谷胱甘肽过氧化物酶1基因的第一条cdna链,使用mint逆转录酶(eurogen,russia)。向6μl的总rna中加入4μl的mint缓冲液、2μl的二硫苏糖醇和2μl的dntp混合物、寡核苷酸gpx1-f和gpx1-r(序列表,(20)和(21))各2μl和2μl的mint逆转录酶,并在42℃下孵育混合物2小时。合成的cdna用作pcr扩增中的基质,其使用相同的寡核苷酸,在94℃下3分钟;30个循环:94℃30秒,60℃30秒和72℃45秒,最终延伸为72℃5分钟。将获得的pcr片段用限制性内切核酸酶bamhi和ecori切割,并与携带卡那霉素抗性基因的3608bp载体连接,并用同样的酶切割。此外,如实施例1所述,从所产生的载体中去除卡那霉素抗性基因。这产生了携带编码人谷胱甘肽过氧化物酶1基因的区域的609-bp的基因治疗dna载体gdtt1.8nas12-gpx1,其允许无抗生素的选择。
实施例4.
获得大肠杆菌菌株jm110-nas,用于生产基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗载体。
通过向其染色体(具体而言向reca基因的区域)中引入线性片段进行同源重组来产生用于工程化基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗载体的大肠杆菌菌株jm110-nas,所述线性片段含有允许无抗生素阳性选择的转座子tn10的调控元件rna-in(64bp),果聚糖蔗糖酶基因sacb,其产物确保在含蔗糖的培养基中进行选择(1422bp),对选择其中发生同源重组的菌株克隆所需的氯霉素抗性基因catr(763bp),以及确保基因reca区域中的同源重组同时基因失活的两个同源序列(同源臂)(左臂和右臂分别为329bp和233bp)。
为了合成左和右同源臂,使用大肠杆菌jm110基因组dna(agilenttechnologies,目录号200239)为基质,通过pcr对基因reca的片段进行扩增。为了合成左同源臂,使用lha-f和lha-r引物(序列表,(22)和(23)),而为了获得右同源臂,使用rha-f和rha-r引物(序列表,(24)和(25))。用in-f、in-1、in-2和in-r合成寡核苷酸(序列表,(26)、(27)、(28)、(29))给rna-in片段加尾。使用b.subtilis168ht基因组dna为基质,以sacb-f和sacb-r为引物(序列表,(30)和(31)),通过pcr扩增产生sacb基因。为了合成catr基因,以大肠杆菌菌株bl21plyss为基质,catr-f和catr-r(序列表,(32)和(33))为引物进行pcr扩增。pcr产物lha(左同源臂)、sacb、rha(右同源臂)如下扩增:94℃3分钟;30个循环:94℃20秒、60℃20秒、72℃60秒,最后延伸72℃5分钟。使用寡核苷酸in-f、in-1、in-2和in-r(序列表,(26)、(27)、(28)和(29))如下合成pcr产物rna-in,94℃3分钟;30个循环:94℃10秒、60℃10秒和72℃10秒以进行片段组装。为此,使用10μm的引物in-f和in-r,和5μm的引物in-1和in-2。根据制造商的说明,使用市售的试剂盒
通过合并5个pcr产物,合成同源重组的线性片段。所有五种产物都有重叠的区域,以允许随后组装成单一片段。将所有片段以10ng等份混合在50μl的体积中。如下衍生pcr产物:94℃3分钟;10个循环:94℃30秒,60℃30秒和72℃2分钟,不加引物。然后加入lha-f、rha-r引物(序列表,(22)、(25)),并再进行25个pcr循环:94℃30秒,60℃30秒和72℃2分钟,最后延伸72℃5分钟。结果,得到了2811-bp长的pcr片段,其具有以下结构:lha-rna-in-sacb-catr-rha。根据制造商的说明,使用dna洗脱试剂盒(biosilica,russia)从琼脂糖凝胶中制备性地回收该片段。
为了获得大肠杆菌菌株jm110-nas,制备了电感受态细胞。为此,使用大肠杆菌菌株jm110(agilenttechnologies)的单个菌落接种10mllb液体培养基,并将细胞在150rpm和37℃的定轨振荡器中培养过夜。第二天,将1/20重新铺板到100mllb液体培养基中,并在定轨振荡器中以150rpm和37℃培养以达到od600=0.5。一旦达到所需的光学密度,将细胞冷却至0℃,在4000g下离心10分钟。接着,取出培养基,用100ml冰冷的双蒸馏水两次洗涤细胞以去除剩余培养基,然后用20ml10%的甘油洗涤。之后,将细胞重悬于1ml的10%甘油中,并用于转化。
通过使用genepulserxcell(bio-rad,usa)在1mm比色杯中以2kv、200ohm、25μf进行电穿孔进行用所获得线性片段的转化。脉冲持续时间在4.9ms至5.1ms。然后将细胞在30℃的定轨振荡器培养箱中在soc培养基中培养2.5小时。然后,将细胞倒入含有10μg/ml氯霉素的lb琼脂化培养基(培养皿)中。细胞在30℃下培养48小时。挑取的克隆在含有酵母提取物、蛋白胨、6%蔗糖和10μg/ml氯霉素的选择性培养基上进行测试存活。所得到的菌株的基因型为recarpsl(strr)thrleuendathi-1lacygalkgaltaratonatsxdamdcmsupe44δ(lac-proab)[f′trad36proablaciqzδm15]cmrsacb+。
实施例5.
获得携带基因载体gdtt1.8nas12和/或基于它的含有治疗性基因的基因治疗dna载体的大肠杆菌菌株jm110-nas/gdtt1.8nas12用于生产基因。
为了制备大肠杆菌株jm110-nas的电感受态细胞,使用单个菌落感染10mllb液体培养基,并以150rpm和37℃在定轨振荡器中培养过夜。第二天,将1/20重新铺板到100ml的lb液体培养基中,在定轨振荡器中以150rpm和37℃培养以达到od600=0.5。达到所需的光学密度时,将细胞冷却至0℃,并在4000g下离心10分钟。接着,取出培养基,并用100ml冰冷的双蒸馏水两次洗涤细胞以去除剩余培养基,然后用20ml10%的甘油洗涤。之后,将细胞重悬于1ml的10%甘油中,并用于通过电穿孔进行的转化。使用genepulserxcell(bio-rad,usa),以2kv,200ohm,25μf在1mm比色杯中进行电穿孔。脉冲的持续时间为4.9至5.1ms,并使用1-10ng的载体。然后将细胞在30℃的定轨振荡器培养箱中在soc培养基中培养2.5小时。然后,将细胞倒入具有含有酵母提取物、蛋白胨、6%蔗糖和10μg/ml氯霉素的选择性培养基的琼脂平板(培养皿)中。由此获得了携带基因治疗dna载体gdtt1.8nas12的大肠杆菌菌株jm110-nas/gdtt1.8nas12。48小时后,使用单个菌落接种于10ml含酵母提取物、蛋白胨、6%蔗糖和10μg/ml氯霉素的液体选择性培养基中,并将培养基以150rpm和37℃在定轨振荡器中培养过夜。第二天,沉淀细胞,并根据制造商的说明,使用genejet质粒微型制备试剂盒(thermofisherscientific)通过碱性裂解提取dna载体。
大肠杆菌菌株jm110-nas/gdtt1.8nas12保存在国家生物资源中心-俄罗斯国家工业微生物典藏(nationalbiologicalresourcecentre–russiannationalcollectionofindustrialmicroorganisms,nbrcrncim,russia)和ncimb专利保存服务机构(uk)的保藏中(注册号vkpm-b-13234,保存日期2018年8月21日;国际保存机构号ncimb43119,保存日期2018年7月19日)。
实施例6.
证明dna载体gdtt1.8nas12的有效性。
为了证实基因治疗dna载体gdtt1.8nas12的有效性,根据实施例2将靶基因例如编码绿色荧光蛋白(gfp)的基因克隆到多接头中。
比较人骨肉瘤细胞mg-63(atcccrl-1427)用质粒载体pefgp-c1(clontech)和基因治疗dna载体gdtt1.8nas12-egfp细胞转染后48小时,治疗性基因例如绿色荧光蛋白(gfp)的积累水平。
为了评估绿色荧光蛋白(gfp)在人骨肉瘤细胞中的积累水平,用质粒载体pefgp-c1(clontech)和基因治疗dna载体gdtt1.8nas12-egfp转染细胞。
在添加有10%牛胚胎血清和10μg/ml庆大霉素的dmem培养基(gibco)中培养mg-63细胞。为了实现90%的汇合,在转染规程前24小时将细胞以4×104个细胞/孔的量接种到24孔板中。lipofectamine3000(thermofisherscientific,usa)用作转染试剂。在试管1中,将1μl质粒载体pefgp-c1和基因治疗dna载体gdtt1.8nas12-egfp溶液(各500ng/μl)和1μl试剂p3000添加到25μl的opti-mem培养基(gibco)。通过轻轻摇动混合制剂。在试管2中,将1μl的lipofectamine3000溶液添加到25μl的培养基opti-mem(gibco)中。通过轻轻摇动混合制剂。将试管1的内容物加入到试管2的内容物中,并在室温下孵育混合物5分钟。将得到的溶液滴加到体积为40μl的细胞中。
使用olympusix53荧光显微镜(japan),用485/535nm滤光片组,48小时后记录结果(图3a)。这些结果证实,用基因治疗dna载体gdtt1.8nas12-egfp转染hek-293细胞,与用质粒载体pefgp-c1(clontech)转染相同的细胞相比,会导致绿色荧光蛋白的积累显著增加。
还通过测量从转染细胞系中提取出的蛋白质的荧光来记录结果。为此,用移液法洗涤孔中的细胞,并以6000rpm10分钟进行沉淀,洗涤两次,然后将沉淀的细胞重悬于1ml磷酸钠缓冲液中。在-70℃下通过三个冻融循环裂解细胞。然后将裂解的细胞的匀浆以13,000g15分钟进行沉淀。将上清液转移到96孔培养板(grainerbio-one)中,每个样品进行四次重复,之后使用fluoroskanascent微孔板荧光仪(labsystems)测量gfp的相对荧光(吸光度455nm/发射538nm)。所得值根据通过bradford蛋白测定法测量的样品中的总蛋白浓度进行标准化。为此,使用考马斯亮蓝r-250(biorad)作为染料。96孔板的孔中的每个重复(每个样品4个重复)用水稀释100倍,之后加入染料。然后使用multiskanascent(thermo)测量所有样品在620nm处的光密度。所得值与用一系列从20到2.5μg/ml的连续稀释液构建的牛血清白蛋白(bio-rad)校准曲线进行比较。使用下式进行计算:
∑蛋白质的量(μ克)={[x]-σ}÷k*m,
其中[x]为每个样品4个重复的od620的平均值,σ为平均偏差,k为bsa的校准曲线的斜率系数,m为样品的稀释系数。
基于从细胞中提取出的蛋白质的总浓度的值,使用下式对样品中的gfp荧光进行标准化:
oen=[oe]÷∑蛋白质的量(mg)
其中
[oe]是每个样品四个重复的平均数,以相对荧光单位。
结果如图3b中所示,其证实用基因治疗dna载体gdtt1.8nas12-egfp转染hek-293细胞,与用质粒载体pefgp-c1(clontech)转染相同细胞相比,增加绿色荧光蛋白的积累水平。
实施例7.
证明dna载体gdtt1.8nas12的有效性。
为了证实基因治疗dna载体gdtt1.8nas12的有效性,根据实施例3将治疗性基因,如体人谷胱甘肽过氧化物酶1(gpx1)基因克隆到多接头中。
用携带人谷胱甘肽过氧化物酶1基因位点的基因治疗dna载体gdtt1.8nas12-gpx1转染48小时后,表皮角质形成细胞heka(pcs-200-011)的原代培养细胞中治疗性基因,例如谷胱甘肽过氧化物酶基因的mrna积累的变化。
使用角质形成细胞生长试剂盒(atccpcs-200-040),在真皮细胞基础培养基(atccpcs-200-030)中培养表皮角质形成细胞heka的原代培养细胞。
为了实现90%的汇合,在转染规程前24小时,将细胞以5×104个细胞/孔的量接种在24孔板中。lipofectamine3000(thermofisherscientific,usa)用作转染试剂。如实施例6中所述用携带人谷胱甘肽过氧化物酶1基因的基因治疗dna载体gdtt1.8nas12-gpx1进行转染。用基因治疗dna载体gdtt1.8nas12转染的hek细胞用作参考。根据实施例3中所述的规程进行从转染细胞提取总rna和构建第一cdna链。为了测量转染后谷胱甘肽过氧化物酶1基因中mrna的表达水平,采用实时pcr(sybrgreenrealtimepcr)方法。为了扩增人gpx1特异性cdna,使用gpx1-sf和gpx1-sr寡核苷酸(序列表,(34),(35))。扩增产物长度为241bp。beta-2微球蛋白(b2m)用作参考基因。
使用quantitectsybrgreenrt-pcr试剂盒(qiagen,usa)或另一种实时pcr试剂盒在20μl的扩增混合物中进行pcr扩增,所述扩增混合物含有:25μlquantitectsybrgreenrt-pcr预混液,2.5mm氯化镁,每个引物0.5μm,和5μl的总rna。对于反应,在以下条件下使用cfx96扩增器(bio-rad,usa):42℃30分钟进行1个循环的逆转录,在98℃15分钟变性,然后进行40个循环,包括在94℃15秒变性,引物在60℃退火30秒,和在72℃30秒进行延伸。阳性对照包括以已知浓度的质粒代表的基质上pcr的扩增子,该质粒含有人谷胱甘肽过氧化物酶1和b2m基因的cdna序列。阴性对照包括去离子水。使用bio-radcfxmanager2.1软件进行pcr产物(即通过扩增获得的gpx-1和b2m基因cdna)的实时定量。
为了证实经携带有人谷胱甘肽过氧化物酶1位点的基因治疗dna载体gdtt1.8nas12-gpx1转染后,表皮角质形成细胞heka的原代培养细胞中谷胱甘肽过氧化物酶1基因表达的增加,图4显示pcr产物积累图。
该图显示,用携带治疗性基因例如人谷胱甘肽过氧化物酶1基因的基因治疗dna载体gdtt1.8nas12-gpx1转染导致人谷胱甘肽过氧化物酶1基因特异性cdna水平大量增加。
实施例8.
证明dna载体gdtt1.8nas12的有效性。
为了证实基因治疗dna载体gdtt1.8nas12的有效性,根据实施例3将治疗性基因例如谷胱甘肽过氧化物酶1基因克隆到多接头中。
测量了将携带治疗性基因(如人谷胱甘肽过氧化物酶1基因)的基因治疗dna载体gdtt1.8nas12-gpx1注入人皮肤时,人皮肤中谷胱甘肽过氧化物酶1蛋白浓度的变化。
为了分析谷胱甘肽过氧化物酶1蛋白浓度的变化,将携带编码谷胱甘肽过氧化物酶1基因的区域的基因治疗dna载体gdtt1.8nas12-gpx1注射到3名患者的前臂皮肤中,同时引入安慰剂,安慰剂是不含谷胱甘肽过氧化物酶1基因cdna的基因治疗dna载体gdtt1.8nas12。患者1,女,66岁(p1);患者2,女,65岁(p2);患者3,男,59岁(p3)。
将携带谷胱甘肽过氧化物酶1基因区域的基因治疗dna载体gdtt1.8nas12(安慰剂)和基因治疗dna载体gdtt1.8nas12-gpx1通过隧道方法用30g针达3mm的深度以每个基因构建体1mg的量进行注射。基因治疗dna载体gdtt1.8nas12(安慰剂)和携带谷胱甘肽过氧化物酶1基因的区域的基因治疗dna载体gdtt1.8nas12-gpx1的注射体积为每个基因构建体1-0.3ml。引入每个基因构建体的点相互之间相距8-10cm。
在注射基因治疗dna载体后的第2天采集活检样品。使用皮肤活检设备epitheasy3.5(medaxsrl)在引入携带编码谷胱甘肽过氧化物酶1基因的基因治疗dna载体gdtt1.8nas12-gpx1(i)、基因治疗dna载体gdtt1.8nas12(安慰剂)(ii)的区域中的患者皮肤,以及从完整的皮肤(iii)取活检样品。患者的皮肤用无菌盐水初步清洗,并用利多卡因溶液麻醉。活检样品体积约为10mm3,且重量约为15mg。将样品置于含有50mmtris-hclph7.6、100mm氯化钠、1mmedta和1mm苯甲基磺酰氟的缓冲溶液中,均质化以获得均匀的悬浮液。所得悬浮液在14,000g离心10分钟。收集上清液,并将其用于使用谷胱甘肽过氧化物酶1(gpx1)的elisa试剂盒(cloud-clonecorp.,usa)按照制造商的方法,用chemwell自动eia和化学分析仪(awarenesstechnologyinc.,usa)进行光密度检测,通过酶联免疫吸附试验(elisa)对治疗性蛋白进行测定。
通过使用来自具有已知浓度的谷胱甘肽过氧化物酶1蛋白的试剂盒的参考样品的校准曲线测定浓度。该方法的灵敏度为5.2ng/ml,测量范围为12.5-200ng/ml。从测定法得到的图如图5中所示。与引入缺乏人谷胱甘肽过氧化物酶1基因的基因治疗dna载体gdtt1.8nas12(安慰剂)的区域中的谷胱甘肽过氧化物酶1蛋白的浓度相比,3名患者各自的皮肤显示在引入携带治疗性基因(例如人谷胱甘肽过氧化物酶1基因)的基因治疗dna载体gdtt1.8nas12的区域中谷胱甘肽过氧化物酶1蛋白的浓度增加。
实施例9.
证明dna载体gdtt1.8nas12的有效性。
为了证实基因治疗dna载体gdtt1.8nas12的有效性,根据实施例2将靶基因,例如编码绿色荧光蛋白(gfp)的基因克隆到多接头中。
比较了用基因治疗dna载体gdtt1.8nas12-egfp进行细胞转染后48小时,牛子宫内膜上皮原代培养细胞(benepc,cellapplications,inc.)中靶基因,例如绿色荧光蛋白(gfp)的积累水平。
为了定量牛子宫内膜上皮原代培养细胞中绿色荧光蛋白(gfp)的积累水平,用基因治疗dna载体gdtt1.8nas12-egfp转染细胞。
根据制造商的说明,使用牛子宫内膜细胞生长培养基试剂盒(cellapplications,inc.)培养细胞。为了时薪90%的汇合,在转染规程前24小时,将细胞以3×104个细胞/孔的量接种在24孔板中。使用lipofectamine3000(thermofisherscientific,usa)作为转染试剂。根据实施例6中所述的规程进行转染。将不含绿色荧光蛋白基因的基因治疗dna载体gdtt1.8nas12用作参考。如实施例6所述,通过测量从转染细胞系中提取的蛋白质的荧光来记录结果。
结果如图中6所示,并允许我们总结为,用携带绿色荧光蛋白基因的基因治疗dna载体gdtt1.8nas12-egfp转染牛子宫内膜上皮原代细胞系,与用缺乏绿色荧光蛋白基因的基因治疗载体gdtt1.8nas12转染相同细胞相比,会导致更高的绿色荧光蛋白的积累水平。
实施例10.
为了证实基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗dna载体的可生产性和可构建性,对大肠杆菌菌株jm110-nas/gdtt1.8nas12进行大规模发酵。
携带基因治疗dna载体gdtt1.8nas12的大肠杆菌菌株jm110-nas/gdtt1.8nas12的发酵在10l的发酵罐中进行,随后提取出基因治疗dna载体gdtt1.8nas12。
为了发酵大肠杆菌菌株jm110-nas/gdtt1.8nas12制备培养基,其每10l含100g胰蛋白胨、50g酵母提取物(bectondickinson),然后用水稀释至8,800ml,并在121℃下高压灭菌20分钟,然后加入1,200ml50%(w/v)的蔗糖。接着,将大肠杆菌菌株jm110-nas/gdtt1.8nas12的种子培养物以100ml的体积接种物接种到培养瓶中。培养物在培养箱振荡器中30℃孵育16小时。将种子培养物转移到techforss生物反应器(inforsht,switzerland)并生长至静止阶段。通过测量600nm处培养物的光密度来控制过程。细胞在5,000-10,000g30分钟沉淀。去除上清液,并将细胞沉淀物重悬于10%体积的磷酸盐缓冲盐水中。将细胞在5,000-10,000g下再次离心30分钟。去除上清液,将20mmtris-hcl、1mmedta、200g/l蔗糖、ph8.0的溶液以1,000ml的体积加入到细胞沉淀物,并充分搅拌混合物,形成均匀的悬浮液。添加蛋溶菌酶溶液至终浓度为100μg/ml。在冰上孵育混合物20分钟,同时轻轻搅拌。接着,加入2,500ml的0.2mnaoh,10g/l十二烷基硫酸钠(sds),在冰上孵育10分钟同时轻轻搅拌,然后加入3,500ml的3m醋酸钠,2m醋酸,ph5-5.5,并将混合物在冰上孵育10分钟同时轻轻搅拌。所得样品在15,000g或更大的值离心20-30分钟。将溶液轻轻倾倒,通过粗滤器(滤纸)过滤除去残留的沉淀。然后加入rnasea(sigma)至终浓度为20ng/ml,并将溶液在室温下孵育16小时过夜。将溶液在15,000g下离心20-30分钟,然后通过0.45μm孔膜过滤器(millipore)。接下来,通过100kda的膜(millipore)进行超滤,并用25mmtris-hcl,ph7.0的缓冲溶液稀释到初始体积。该操作进行三到四次。将该溶液施加于具有250mldeaesepharosehp的柱(ge,usa),用25mmtris-hcl,ph7.0平衡。样品施加后,用三个体积的相同溶液洗涤柱,然后用25mmtris-hcl、ph7.0线性梯度洗脱基因dna载体gdtt1.8nas12以获得25mmtris-hcl、ph7.0、1mnacl的溶液(柱体积的5倍)。洗脱过程通过测量260nm处的径流溶液的光密度来控制。将含有基因治疗dna载体gdtt1.8nas12的层析级分合并,并通过superdex200吸附剂(ge,usa)进行凝胶过滤。用磷酸盐缓冲盐水平衡柱。通过测量260nm处的径流溶液的光密度控制洗脱过程,并通过琼脂糖凝胶电泳分析各级分。合并含有基因治疗dna载体gdtt1.8nas12的级分,并在-20℃下保存。为了评估该过程的可重复性,将这些过程操作重复三次。过程的可重复性和最终产品产量的定量特征证实了基因治疗dna载体gdtt1.8nas12和/或基于它的携带治疗性基因的基因治疗dna载体在工业规模上的可生产性和可构建性。
因此,本发明的目的,具体而言,构建用于人和动物细胞遗传修饰的多功能基因治疗dna载体将合理地组合:
i)由于不超过2600bp,即2591bp的有限的长度,确保有效地渗透到靶细胞中,以及调控元件序列的可用性,确保在大多数人和动物组织细胞中治疗性基因的高表达,基因治疗dna载体对提高人和动物组织细胞中治疗性基因的表达水平的有效性。
ii)由于基因治疗dna载体中不存在代表病毒基因组的核苷酸序列的调控元件,安全应用于人类和动物的基因治疗的可能性。
iii)由于基因治疗dna载体中不存在抗生素抗性基因,安全应用于人和动物的基因治疗的可能性。
iv)基因治疗dna载体和/或基于它的携带治疗性基因的基因治疗dna载体的在工业规模上的可生产性和可构建性。
已经实现,其通过以下实施例得以支持:对于项(i),实施例1、6、7、8、9;对于项(ii),实施例1;对于项(iii),实施例1;对于项(iv),实施例4、5、10。
工业适用性
以上实施例均证实了所提出的基因治疗dna载体gdtt1.8nas12,用于获得其的方法,用于生产dna载体gdtt1.8nas12的大肠杆菌菌株jm110-nas,获得大肠杆菌菌株jm110-nas、包括基因治疗dna载体gdtt1.8nas12的大肠杆菌菌株jm110-nas/gdtt1.8nas12用于其生产的方法,获得大肠杆菌菌株jm110-nas/gdtt1.8nas12的方法以及基因治疗dna载体的工业规模生产的方法的工业适用性。
缩略语清单
gdtt1.8nas12是缺乏病毒基因组序列和抗生素抗性标记的基因治疗载体。
dna—脱氧核糖核酸
cdna—互补性脱氧核糖核酸
rna—核糖核酸
mrna—信使核糖核酸
bp—碱基对
pcr—聚合酶链反应
ml—毫升,μl—微升
l—升
μg—微克
mg—毫克
g—克
μm—微摩尔
mm—毫摩尔
min—分钟
s—秒
rpm—每分钟转数
nm—纳米
cm—厘米
mw—毫瓦
r.u.f—相对荧光单位
pbs—磷酸盐缓冲盐水
序列表
<110>基因诊断与治疗21有限公司,obschestvosogranichennoiotvetstvennostju?rekombitekh?
<120>基因治疗dna载体gdtt1.8nas12及用于获得它的方法
<160>2
<170>bissap1.3.6
<210>1
<211>2591
<212>dna
<213>智人
<400>1
ataacgcaggaaagaacatgtgagcaaaaggccagcaaaaggccagggaccgtaaaaagg60
ccgcgttgctggcgtttttccataggctccgcccccctgacgagcatcacaaaaatcgac120
gctcaagtcagaggtggcgaaacccgacaggactataaagataccaggcgtttccccctg180
gaagctccctcgtgcgctctcctgttccgaccctgccgcttaccggatacctgtccgcct240
ttctcccttcgggaagcgtggcgctttctcatagctcacgctgtaggtatctcagttcgg300
tgtaggtcgttcgctccaagctgggctgtgtgcacgaaccccccgttcagcccgaccgct360
gcgccttatccggtaactatcgtcttgagtccaacccggtaagacacgacttatcgccac420
tggcagcagccactggtaacaggattagcagagcgaggtatgtaggcggtgctacagagt480
tcttgaagtggtggcctaactacggctacactagaagaacagtatttggtatctgcgctc540
tgctgaagccagttaccttcggaaaaagagttggtagctcttgatccggcaaacaaacca600
ccgctggtagcggtggtttttttgtttgcaagcagcagattacgcgcagaaaaaaaggat660
ctcaagaagatcctttgatcttttctacctcgaccgtgaggctccggtgcccgtcagtgg720
gcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgaacc780
ggtgcctagagaaagtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgc840
ctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctt900
tttcgcaacgggtttgccgccagaacacaggtaagtgccgtgtgtggttcccgcgggcct960
ggcctctttacgggttatggcccttgcgtgccttgaattacttccacgcccctggctgca1020
gtacgtgattcttgatcccgagcttcgggttggaagtgggtgggagagttcgaggccttg1080
cgcttaaggagccccttcgcctcgtgcttgagttgaggcctggcctaggcgctggggccg1140
ccgcgtgcgaatctggtggcaccttcgcgcctgtctcgctgctttcgataagtctctagc1200
catttaaaatttttgatgacctgctgcgacgctttttttctggcaagatagtcttgtaaa1260
tgcgggccaagatctgcacactggtatttcggtttttggggccgcgggcggcgacggggc1320
ccgtgcgtcccagcgcacatgttcggcgaggcggggcctgcgagcgcggccaccgagaat1380
cggacgggggtagtctcaagctggccggcctgctctggtgcctggcctcgcgccgccgtg1440
tatcgccccgccctgggcggcaaggctggcccggtcggcaccagttgcgtgagcggaaag1500
atggccgcttcccggccctgctgcagggagctcaaaatggaggacgcggcgctcgggaga1560
gcgggcgggtgagtcacccgcacaaaggaaaagggcctttccgtcctcagccgtcgcttc1620
atgtgactccacggagtaccgggcgccgtccaggcacctcgattagttctcgagcttttg1680
gagtacgtcgtctttaggttggggggaggggttttatgcgatggagtttccccacactga1740
gtgggtggagactgaagttaggccagcttggcacttgatgtaattctccttggaatttgc1800
cctttttgagtttggatcttggttcattctcaagcctcagacagtggttcaaagtttttt1860
tcttccatttcaggtgtcgtgaaaactacccctaaaagccaggatccgatatcgtcgaca1920
agcttggtacctccggagcggccgctctagagctagcgacgtcgaattccctgtgacccc1980
tccccagtgcctctcctggccctggaagttgccactccagtgcccaccagccttgtccta2040
ataaaattaagttgcatcattttgtctgactaggtgtccttctataatattatggggtgg2100
aggggggtggtatggagcaaggggcaagttgggaagacaacctgtagggcctgcggggtc2160
tattgggaaccaagctggagtgcagtggcacaatcttggctcactgcaatctccgcctcc2220
tgggttcaagcgattctcctgcctcagcctcccgagttgttgggattccaggcatgcatg2280
accaggctcagctaatttttgtttttttggtagagacggggtttcaccatattggccagg2340
ctggtctccaactcctaatctcaggtgatctacccaccttggcctcccaaattgctggga2400
ttacaggcgtgaaccactgctcccttccctgtccttacgcgtagaattggtaaagagagt2460
cgtgtaaaatatcgagttcgcacatcttgttgtctgattattgatttttggcgaaaccat2520
ttgatcatatgacaagatgtgtatctaccttaacttaatgattttgataaaaatcattaa2580
ctagtccatgg2591
<210>2
<211>609
<212>dna
<213>智人
<400>2
atgtgtgctgctcggctagcggcggcggcggcggcggcccagtcggtgtatgccttctcg60
gcgcgcccgctggccggcggggagcctgtgagcctgggctccctgcggggcaaggtacta120
cttatcgagaatgtggcgtccctctgaggcaccacggtccgggactacacccagatgaac180
gagctgcagcggcgcctcggaccccggggcctggtggtgctcggcttcccgtgcaaccag240
tttgggcatcaggagaacgccaagaacgaagagattctgaattccctcaagtacgtccgg300
cctggtggtgggttcgagcccaacttcatgctcttcgagaagtgcgaggtgaacggtgcg360
ggggcgcaccctctcttcgccttcctgcgggaggccctgccagctcccagcgacgacgcc420
accgcgcttatgaccgaccccaagctcatcacctggtctccggtgtgtcgcaacgatgtt480
gcctggaactttgagaagttcctggtgggccctgacggtgtgcccctacgcaggtacagc540
cgccgcttccagaccattgacatcgagcctgacatcgaagccctgctgtctcaagggccc600
agctgtgcc609
寡核苷酸序列表:
(1)寡核苷酸ucori-bamggatccagatctactcgaggtagaaaagatcaaaggatcttc
(2)寡核苷酸ucori-ncoagtccatggataacgcaggaaagaacatgtg
(3)寡核苷酸hgh-faggatccgaattccctgtgacccctccccag
(4)寡核苷酸hgh-rctctttaccaattctacgcgtaaggacagggaagggagca
(5)寡核苷酸ro-fcttccctgtccttacgcgtagaattggtaaagagagtcgt
(6)寡核苷酸ro-rccgtagaaaactagttaatgatttttatcaaaatcattaag
(7)寡核苷酸ro-1gaattggtaaagagagtcgtgtaaaatatcgagttcgcacatcttgttg
(8)寡核苷酸ro-2gatttttggcgaaaccatttgatcatatgacaagatgtgtatctacc
(9)寡核苷酸ro-3atgatttttatcaaaatcattaagttaaggtagatacacatcttgtc
(10)寡核苷酸kan-faaatcattaactagttttctacggggtctgacgc
(11)寡核苷酸kan-rcagccatggactagtggtggcacttttcgggga
(12)寡核苷酸mcs1gatccgatatcgtcgacaagcttggtacct
(13)寡核苷酸mcs2caagcttgtcgacgatatcg
(14)寡核苷酸mcs3ccggagcggccgctctagagctagcgacgtcg
(15)寡核苷酸mcs4aattcgacgtcgctagctctagagcggccgctccggaggtac
(16)寡核苷酸ef1-xhoatctcgagcgtgaggctccggtgcc
(17)寡核苷酸ef1-rtcggatcctggcttttaggggtagttttc
(18)寡核苷酸mvgfp-fgggatccaccggtcgcca
(19)寡核苷酸mvgfp-ratagaattcttacttgtacagctcgtcca
(20)寡核苷酸gpx1-fgggatccatgtgtgctgctcggc
(21)寡核苷酸gpx1-ratagaattcttaggcacagctgggatt
(22)寡核苷酸lha-fgctgacgctgcaggtgatc
(23)寡核苷酸lha-rgacaagatgtgtgtctaccgcttcaggttacccgccag
(24)寡核苷酸rha-ftggcagggcggggcgtaactacgcctctgttcgtctcga
(25)寡核苷酸rha-rctcagcagcaactcacgtac
(26)寡核苷酸in-fctggcgggtaacctgaagcggtagacacacatcttgtc
(27)寡核苷酸in-1atttttggcgaaaccattctatcatatgacaagatgtgtgtc
(28)寡核苷酸in-2atatgatagaatggtttcgccaaaaatcaataatcagacaac
(29)寡核苷酸in-rcaaactttttgatgttcatcttgttgtctgattattg
(30)寡核苷酸sacb-fcaataatcagacaacaagatgaacatcaaaaagtttg
(31)寡核苷酸sacb-rcttacgtgccgatcattatttgttaactgttaattgtc
(32)寡核苷酸catr-fcaattaacagttaacaaataatgatcggcacgtaagagg
(33)寡核苷酸catr-rcgagacgaacagaggcgtagttacgccccgccctgccac
(34)寡核苷酸gpx1-sfggcaccacggtccgggactac
(35)寡核苷酸gpx1-srcagggcctcccgcaggaaggc
- 还没有人留言评论。精彩留言会获得点赞!