来自单个生物样品的蛋白质、核糖体和无细胞核酸的同时基于测序的分析的制作方法
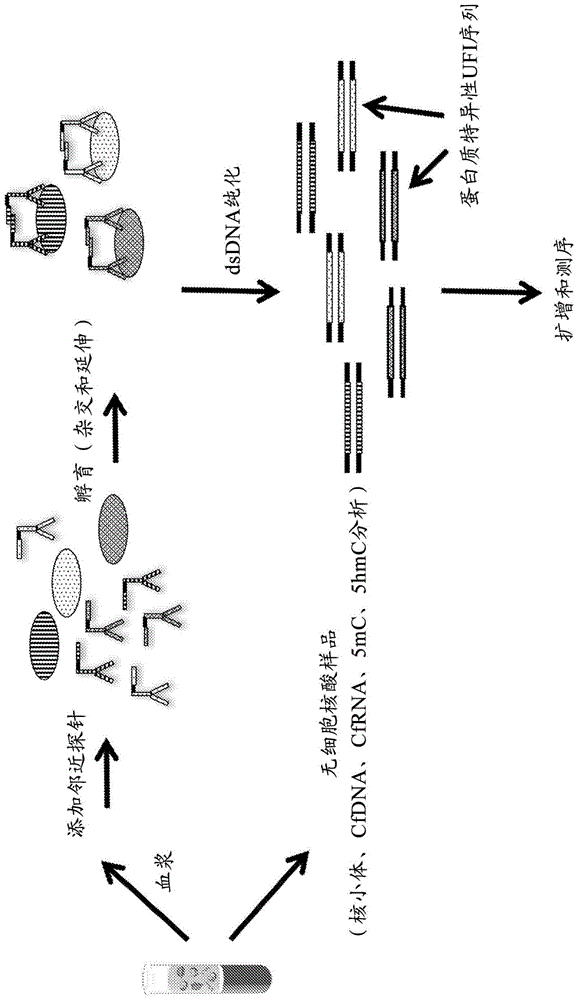
本发明一般地涉及表观遗传学分析,并且更具体地涉及用于从单个生物样品获得多种类型信息的组合工作流程方法。本发明发现可用于基因组学、医学、诊断学和表观遗传学研究的领域。技术背景从包含微量分析物的相对较小的生物样品中获取大量信息具有独特的挑战。例如,无细胞dna(cfdna)样品通常每毫升血浆仅包含几纳克的dna。结果是,难以评估无细胞dna样品的多于一个或两个特征,例如dna序列信息和/或甲基化数据,每个特征经常使用单独的工作流程,将已少量的dna样品分开作为输入,并且限制了可获知的关于同一起始分子的信息量(例如,如果单个起始cfdna片段模板包含甲基化细胞因子和羟甲基化细胞因子二者)。然而,已经提出了一种用于从一个cfdna样品中获得不同类型信息的方法。参见,例如,arensdorf等的于2018年2月14日提交的临时美国专利申请序列号62/630,798的“methodsfortheepigeneticanalysisofdna,particularlycell-freedna”(bluestargenomics,inc.),其描述了一种用于检测单个无细胞dna样品中的不同表观遗传学特征的方法,包括dna片段的5-甲基胞嘧啶(5mc)和5-羟甲基胞嘧啶(5hmc)残基的存在和位置,其中最终将经差异加工的dna片段合并并一起测序以提供期望的信息;以及song等的美国专利公开号2017/0298422a1的“simultaneoussingle-moleculeepigeneticimagingofdnamethylationandhydroxymethylation”(小利兰·斯坦福大学董事会),其描述了将不同的标记与无细胞dna的5mc和5hmc结合,然后检测和分析通过标记产生的信号。还参见reuter等的专利公开号2018/0080021的“simultaneoussequencingofrnaanddnafromthesamesample”,其涉及一种从单个生物样品中扩增和测序rna和dna二者的方法。如果可以从单个小体积生物样品中获得多得多的信息,这将是极其有用的,包括但不限于关于dna序列、dna表观遗传学修饰、rna序列、核小体结构和定位、组蛋白修饰以及核酸相关和游离血浆蛋白二者的信息。此外,如果此种全面的数据集可以以基本上相同的方式并在组合工作流程的背景下从单个小体积生物样品中产生,无需并行加工、另外的样品材料或多种信息产生方法论,这将是理想的。最后,到可以将此种非序列信息一起编码(例如在序列中,但使用标准的未经修饰的核酸碱基)在同一模板核酸中,并与原始亲本模板序列一起通过后续步骤的加工的程度,这将增强与现有研究工作流程和二代测序文库制备技术的兼容性。最具体地,这将使得能够使用提取、扩增和检测技术,这些提取、扩增和检测技术原本可从相关序列中稀释或分离此种非序列信息。技术实现要素:本发明针对本领域中的前述需求,并且在一个实施方案中,提供了一种用于分析生物样品以测定其中的多种类型信息的组合工作流程方法,无需许多独立的分析步骤、多种数据生成方式或大量样品样品。可以从患者的血液样品中获得的信息类型包括例如特定血浆蛋白的存在和浓度;与cfdna(例如血液样品中无细胞级分的dna)相关的组蛋白修饰的数量、位置和类型;该级分中cfrna和cfdna的序列;关于无细胞dna的表观遗传学信息,例如羟甲基化和甲基化图谱,即分别地5-羟甲基胞嘧啶(5hmc)和5-甲基胞嘧啶(5mc)残基的分布。本发明另外地关于用于分析生物样品以测定样品的一个或更多个非经典序列特征的基于经典测序的方法,其中“非经典序列特征”是指除了同一性和样品中核酸分子的四个主要碱基的顺序(即,dna的腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶,rna的腺嘌呤、胞嘧啶、鸟嘌呤和尿嘧啶)之外的特征。即,该方法包括测定经典核酸序列信息,可从中衍生出目标非经典序列特征。非经典序列特征可以是与核酸的组成有关的信息,例如经修饰的胞嘧啶残基的分布,例如5hmc或5mc,或者它可以与核酸的组成无关,而是关于血液样品中血浆蛋白的存在和浓度、在血液样品中无细胞核小体级分中观察到的组蛋白修饰等。可以执行该方法来测定生物样品的单个非经典序列特征,生物样品的多于一个非经典序列特征、或经典序列信息和一个或更多个非经典序列特征的组合。该分析涉及将目标非经典序列特征的转换成经典序列数据,例如血浆蛋白的鉴定,血浆蛋白的浓度,组蛋白修饰的数量、位置和类型,核酸的羟甲基化图谱(例如生物样品的无细胞核酸部分中无细胞dna的5hmc图谱),或核酸的甲基化图谱(例如,生物样品的无细胞核酸级分中无细胞dna的5mc图谱)。获得的经典序列数据包括长度为约4至约36个碱基对的至少一种特定核酸序列,其用作独特特征标识符(ufi)序列,其中将ufi掺入衍生自生物样品中的目标分析物的双链dna(dsdna)分子中。经典序列数据还可以包含cdna序列,从而提供有关rna模板分子的相应序列的信息,例如生物样品的无细胞核酸级分中的无细胞rna。在第一实施方案中,本发明提供了一种改进的邻近延伸测定法,其通过提供多种探针对来鉴定生物样品中的多种蛋白质分析物,每种探针对包含第一邻近探针和第二邻近探针,其中每种探针对靶向特定蛋白质分析物,并且在相应的蛋白质分析物的存在下在每种探针对的探针之间产生双链dna(dsdna)片段,其所述改进包括:(a)将用作蛋白质标识符条形码的蛋白质特异性核酸序列掺入所述双链dsdna片段中,从而形成蛋白质条形码化的dsdna模板分子;(b)对所述蛋白质条形码化的dsdna模板分子进行扩增和测序;和(c)从产生的序列读取中观察到的所述蛋白质标识符条形码中鉴定所述生物样品中的蛋白质分析物。在一些实施方案中,每个蛋白质特异性核酸序列包含在衔接子内,并且步骤(a)包括将所述衔接子与所述dsdna片段末端连接。在一个相关实施方案中,提供了一种改进的邻近延伸测定法,其通过提供多种探针对来鉴定生物样品中的多种蛋白质分析物,每种探针对包含第一邻近探针和第二邻近探针,其中每种探针对靶向特定蛋白质分析物,并且在相应的蛋白质分析物的存在下在每种探针对的探针之间产生dsdna片段,其中所述改进包括:(a)将以下项掺入所述双链dna片段:(i)用作蛋白质标识符条形码的蛋白质特异性核酸序列,和(ii)包含5hmc残基的捕获序列,从而形成包含所述捕获序列的蛋白质条形码化的dsdna模板分子;(b)对所述蛋白质条形码化的dsdna模板分子进行扩增和测序;和(c)从产生的序列读取中观察到的所述蛋白质标识符条形码中鉴定所述生物样品中的蛋白质分析物。在一些实施方案中,步骤(a)通过将所述dsdna片段与衔接子末端连接来进行,所述衔接子各自包含蛋白质特异性核酸序列和所述捕获序列。事实上,可以将捕获序列中的一个或更多个5hmc残基官能化,以促进从样品、样品级分或包含多种生物分子的混合物中去除dsdna模板分子。这在对单个生物样品进行组合工作流程分析(从中提取多种类型信息)的情况下特别有用。在另一个实施方案中,提供了一种改进的邻近延伸测定法,其通过提供多种探针对来鉴定生物样品中的多种蛋白质分析物,每种探针对包含第一邻近探针和第二邻近探针,其中每种探针对靶向特定蛋白质分析物,并且在相应的蛋白质分析物的存在下在每种探针对的探针之间产生dsdna片段,其中所述改进包括:(a)将以下项掺入所述双链dna片段:(i)用作蛋白质识别条形码的蛋白质特异性核酸序列,(ii)用作分子条形码的随机核酸序列,和任选地(iii)包含5hmc残基的捕获序列,从而形成任选地包含所述捕获序列的蛋白质条形码化的dsdna模板分子;(b)对所述蛋白质条形码化的dsdna模板分子进行扩增和测序;和(c)从产生的序列读取中观察到的所述蛋白质标识符条形码中鉴定所述生物样品中的蛋白质分析物。在一个相关实施方案中,所述邻近性延伸测定法还包括在步骤(b)之前,将至少一种蛋白质浓度对照组合物与所述dsdna模板分子组合。对照组合物与分子条形码一起能够通过将指示特定蛋白质分析物的序列读取的数目与由蛋白质浓度对照组合物产生的序列读取比较来测定样品中至少一种蛋白质分析物的原始浓度。在另一个实施方案中,提供了一种改进的邻近延伸测定法,其用于鉴定多个生物样品中每一个中的多种蛋白质分析物,其中,对于每个生物样品,所述测定法包括提供多种探针对,每种探针对包含第一邻近探针和第二邻近探针,其中每种探针对靶向特定的蛋白质分析物,并且在相应的蛋白质分析物的存在下在每种探针对的探针之间产生双链dna片段,其中所述改进包括:(a)将以下项掺入所述双链dna片段:(i)用作蛋白质识别条形码的蛋白质特异性核酸序列,任选地(ii)用作分子条形码的随机核酸序列,和任选地(iii)包含5hmc残基的捕获序列,从而形成任选地包含所述捕获序列的蛋白质条形码化的dsdna模板分子;(b)对所述蛋白质条形码化的dsdna模板分子进行扩增和测序;(c)从产生的序列读取中观察到的所述蛋白质标识符条形码中鉴定所述生物样品中的蛋白质分析物;和(d)对至少100个生物样品并行进行步骤(a)、(b)和(c)。例如,可以对至少300个生物样品、至少500个生物样品或至少1500个生物样品同时进行步骤(a)、(b)和(c)。在一个相关实施方案中,本发明提供了一种使用基于dna序列的技术鉴定生物样品中的多种蛋白质分析物的方法,所述方法包括:(a)提供多种探针对,每种探针对靶向特定蛋白质分析物,并且包含在第一末端的蛋白质结合结构域、在相对的第二末端的核酸结合结构域和在其之间的非杂交核酸区域,其中(i)探针对的第一邻近探针和第二邻近探针的蛋白质结合结构域能够同时与同一蛋白质分析物上的不同结合位点结合,并且(ii)所述探针的核酸结合结构域彼此互补并且当所述第一邻近探针和所述第二邻近探针都与蛋白质结合并且足够邻近以发生杂交时杂交以形成dsdna片段;(b)在有效促进以下项的条件下用所述探针对孵育所述生物样品或其级分:(i)探针对中每个邻近探针的蛋白质结合结构域与相应的蛋白质分析物结合和(ii)核酸结合结构域彼此杂交以形成dsdna片段,所述dsdna片段具有源自所述第一邻近探针的5'末端和源自所述第二邻近探针的3'末端;(c)通过添加聚合酶和dntp的混合物将第一邻近探针的3'末端沿着第一邻近探针延伸,以在所述探针之间产生dsdna片段,所述dsdna片段掺入用作蛋白质标识符条形码的蛋白质特异性核酸序列和包含5hmc残基的捕获序列,其中(i)所述第一探针、所述第二探针或所述第一探针和所述第二探针二者的核酸结合区域包含所述捕获序列、所述蛋白质标识符条形码或所述捕获序列和所述蛋白质标识符条形码二者;(ii)所述dntp的混合物包含至少一种5hmc残基;和/或(iii)在聚合酶延伸后将衔接子连接至所述dsdna片段的末端,其中至少一个衔接子包含所述捕获序列、所述蛋白质标识符条形码或所述捕获序列和所述蛋白质标识符条形码二者,从而形成蛋白质-条形码化的dsdna模板分子,每个dsdna模板分子包含捕获序列;(d)对包含捕获序列的蛋白质条形码化的dsdna模板分子进行扩增和测序;以及(e)从步骤(b)中产生的序列读取中观察到的蛋白质标识符条形码中鉴定所述生物样品中的蛋白质分析物。在前述实施方案的一方面,所述方法在生物样品的级分上进行,典型地在从血液样品获得的血浆上进行。在一个另外实施方案中,提供了一种组合工作流程方法,其中如关于上述实施方案中任一个所阐释的分析一个或更多个生物样品中的蛋白质分析物,以及分析来自同一生物样品的无细胞核酸样品。在本文提供的组合工作流程方法的第一实施方案中,针对无细胞核酸样品获得的信息关于无细胞核酸样品中的核小体内的一种或更多种组蛋白修饰的存在或数量。组蛋白修饰可以是共价翻译后修饰(ptm),影响基因表达的组蛋白结构改变。在该实施方案的一方面,具体的目标组蛋白修饰是用于评估受试者的疾病状态的组蛋白修饰生物标志物,也可以包括用于评估受试者的疾病状态的组蛋白修饰生物标志物。在本文提供的组合工作流程方法的另一个实施方案中,针对无细胞核酸样品获得的信息包括无细胞核酸样品中的至少一种cfdna序列。在本文提供的组合工作流程方法的另一个实施方案中,针对无细胞核酸样品获得的信息包括无细胞核酸样品中的至少一种cfdna序列。在本文提供的组合工作流程方法的另一个实施方案中,针对无细胞核酸样品获得的信息包括关于cfdna羟甲基化的表观遗传学数据。在本文提供的组合工作流程方法的另一个实施方案中,针对无细胞核酸样品获得的信息包括关于cfdna甲基化的表观遗传学数据。在另一个实施方案中,提供了一种组合工作流方法,其中如上所述的分析一个或更多个生物样品中的蛋白质分析物,并且分析同一生物样品的无细胞核酸样品的以下项中的至少两种:组蛋白修饰、cfdna序列、cfrna序列、cfdna羟甲基化和cfdna甲基化。在另一个实施方案中,本发明提供了一种使用基于dna测序的技术制备无细胞核酸样品以能够鉴定其中所含核小体中的至少一种组蛋白修饰的方法。所述方法包括:(a)提供无细胞核酸样品,其包含多种核小体,每种核小体包含缠绕在组蛋白核心周围的cfdna分子;(b)将包含末端杂交区域的衔接子连接至所述cfdna分子的末端,从而提供经修饰的无细胞核酸样品,其包含各自缠绕在组蛋白核心周围的衔接子-连接的cfdna分子;(c)提供邻近探针,所述邻近探针包含:在第一末端的与目标组蛋白修饰特异性结合的组蛋白修饰结合结构域;在第二末端的与末端杂交区域互补的核酸结合结构域;和在其之间的非杂交区域,其包含与目标组蛋白修饰相对应并且从而用作组蛋白修饰条形码的核酸序列,其中所述邻近探针的大小设置成同时允许所述组蛋白修饰结合结构域与所述目标组蛋白修饰结合并且互补核酸结合结构域与所述杂交核酸区域杂交;(d)在有效促进以下项的条件下用所述邻近探针孵育所述经修饰的无细胞核酸样品:(i)所述组蛋白修饰结合结构域与所述组蛋白修饰结合和(ii)所述互补核酸结合结构域与所述杂交核酸区域杂交以形成dsdna片段,所述dsdna片段具有源自所述无细胞dna的5'末端和源自所述邻近探针的3'末端并且包含所述组蛋白修饰条形码;以及(e)通过添加聚合酶和dntp的混合物将所述dsdna片段的5'末端沿着所述邻近探针的非杂交区域和所述组蛋白修饰条形码延伸,从而提供用于扩增和测序的组蛋白修饰条形码化的dsdna模板分子。在一个相关实施方案中,步骤(c)包括提供多种邻近探针,每种邻近探针靶向不同的组蛋白修饰。在另一个相关实施方案中,所述方法另外地包括对组蛋白修饰条形码话的dsdna模板分子进行扩增。在另一个相关实施方案中,所述方法还包括对扩增的组蛋白修饰条形码化的dsdna模板分子进行测序,并且从产生的序列读取中观察到的组蛋白修饰条形码中测定有关组蛋白修饰的类型和位置的信息。本发明的另一个实施方案关于一种在制备用于从无细胞核酸样品中提取的cfdna中使用包含至少一个5hmc残基的衔接子的方法。所述方法包括(a)将包含捕获序列的dna衔接子连接至所述无细胞核酸样品中的末端钝化dna的末端,以提供连接有衔接子的dna,所述dna捕获序列包含5hmc残基;以及(b)用亲和标签将所述5hmc残基官能化,所述亲和标签允许选择性去除标签化的cfdna。亲和标签可以是生物素部分,例如生物素本身,或更通常是已经被共价修饰以包括反应位点的生物素。然后生物素化的5hmc位点用于通过与亲和素包被或链霉亲和素包被的支撑物反应实现从样品中提取。在前述实施方案的一方面,衔接子另外地包含ufi序列,通常至少两种ufi序列,每种ufi序列指示无细胞核酸序列中cfdna的非序列特征或特点。在扩增和测序之后,可以从序列读取中观察到的ufi序列测定目标非序列特征。在另一个实施方案中,本发明提供了一种在单个无细胞核酸样品中制备无细胞dna和无细胞rna用以同时进行基于测序的分析的方法。所述方法(a)包括将包含第一衔接子序列的dna衔接子连接至所述无细胞样品中的末端钝化dna的末端以提供连接有衔接子的dna,所述第一衔接子序列包含至少一种ufi序列,其中所述至少一种ufi序列包含源标识符条形码;(b)将所述连接有衔接子的dna和rna纯化,以提供所述连接有衔接子的dna和rna的无细胞混合物;(c)从所述rna合成cdna的第一链;(d)合成cdna的与所述第一链互补的第二链以提供cdna双链体;以及(e)在不存在连接酶的情况下,将包含第二衔接子序列的cdna衔接子共价附接至所述cdna双链体的至少一个末端,所述第二衔接子序列包含源标识符条形码和rna指示剂条形码,从而在无细胞混合物中提供衔接子结合的cdna,所述无细胞混合物还包含所述连接有衔接子的dna。在本发明的另一个实施方案中,提供了一种组合工作流程方法,其用于使用基于测序的分析从单个无细胞核酸样品中提取多种类型数据,其中所述数据包括样品中cfrna的羟甲基化图谱以及cfrna的序列信息。所述数据还可以包括dna序列信息。所述方法包括:(a)将包含第一衔接子序列的dna衔接子连接至所述无细胞核酸样品中的末端钝化cfdna的末端上以提供连接有衔接子的dna,所述第一衔接子序列包含至少一种ufi序列,其中至少一种ufi序列包含源标识符条形码;(b)从所述样品中的rna合成cdna,并且将包含所述源标识符条形码和rna指示剂条形码的cdna衔接子共价附接至所述cdna的至少一个末端,从而在无细胞组合物中提供衔接子结合的cdna,所述无细胞组合物还包含所述连接有衔接子的dna;(c)用亲和标签将无细胞组合物中的5hmc残基官能化,所述亲和标签允许从所述无细胞组合物中选择性去除含5hmc的dna;(d)从所述无细胞组合物中去除所述含5hmc的dna,其中未标签化的dna和衔接子结合的cdna保留;(e)将5hmc过程条形码附加到所述含5hmc的dna;以及(f)将条形码化的含5hmc的dna、所述未标签化的dna和所述衔接子结合的cdna合并、扩增和测序。在前述实施方案的一方面,步骤(e)通过将所述5hmc过程条形码掺入所述dna衔接子进行。在本发明的一个相关实施方案中,提供了一种组合工作流程方法,其用于使用基于测序的分析从单个无细胞核酸样品中提取多种类型数据,其中所述数据包括样品中cfdna的羟甲基化谱、cfrna序列信息和任选地dna序列信息,如上所述,并且还包括样品中cfdna的甲基化图谱。所述方法包括:(a)将包含第一衔接子序列的dna衔接子连接至所述无细胞核酸样品中的末端钝化cfdna的末端上以提供连接有衔接子的dna,所述第一衔接子序列包含至少一种ufi序列,其中所述至少一种ufi序列包含源标识符条形码;(b)从所述样品中的rna合成cdna,并且将包含所述源标识符条形码和rna指示剂条形码的cdna衔接子共价附接至所述cdna的至少一个末端,从而在无细胞组合物中提供衔接子结合的cdna,所述无细胞组合物还包含所述连接有衔接子的dna;(c)用亲和标签将所述无细胞组合物中的5hmc残基官能化,所述亲和标签允许从所述无细胞组合物中选择性去除含5hmc的dna;(d)从所述无细胞组合物中去除所述含5hmc的dna,其中未标签化的dna和衔接子结合的cdna保留;(e)将5hmc过程条形码附加到所述含5hmc的dna;(f)将剩余样品中的甲基胞嘧啶残基转化为氧化的甲基胞嘧啶残基;(g)用第二亲和标签将所述氧化的甲基胞嘧啶残基官能化,所述第二亲和标签允许从所述样品中选择性去除官能化的物质;(h)去除所述标签化的含5mc的dna,其中未标签化的dna和衔接子结合的cdna保留在所述样品中;(i)将5mc过程条形码附加到所述标签化的含5mc的dna;以及(j)对所述标签化的含5hmc的dna、所述标签化的含5mc的dna、所述未标签化的dna和所述衔接子结合的cdna进行扩增和测序。在本发明的另一个相关实施方案中,提供了一种组合工作流程方法,其用于使用基于测序的分析从单个无细胞核酸样品中提取至少两种类型的数据,其中所述数据包括样品中的cfdna的羟甲基化图谱、cfrna序列信息和任选地dna序列信息。所述方法包括:(a)将包含第一衔接子序列的dna衔接子连接至所述样品中的末端钝化dna的末端上以提供连接有衔接子的dna,所述第一衔接子序列包含至少一种分子条形码,所述至少一种分子条形码包含源标识符条形码;(b)从所述样品中的rna合成cdna,并且将包含5hmc残基、源标识符条形码和rna指示剂条形码的cdna衔接子共价附接至所述cdna的至少一个末端,从而提供条形码化的衔接子结合的cdna;(c)用亲和标签将所述样品中的5hmc残基官能化,所述亲和标签允许从所述无细胞样品中选择性去除含5hmc的物质;(d)从所述无细胞样品中去除所述含5hmc的dna和所述条形码化的衔接子结合的cdna;以及(e)对所述含5hmc的dna和所述条形码化的衔接子结合的cdna的合并混合物进行扩增和测序以提供同一样品中的关于dna羟甲基化和cfrna的数据。在本发明的另一个相关实施方案中,提供了一种组合工作流程方法,用于使用基于测序的分析从单个无细胞核酸样品中提取至少两种类型的数据,其中所述数据包括无细胞核酸样品中的核小体内一种或更多种组蛋白修饰的存在或数量以及样品中cfrna的序列信息。所述方法包括:一种用于从单个无细胞核酸样品中提取多种类型数据的组合工作流程方法,其包括:(a)将包含杂交核酸区域的衔接子连接至核小体相关dna的每个末端,从而提供包含与连接有衔接子的dna相关的核小体的经修饰的无细胞核酸样品;(b)提供邻近探针,所述邻近探针包含在第一末端的组蛋白修饰结合结构域、在相对的第二末端的与所述杂交核酸区域互补的核酸结合结构域和在其之间的非杂交区域,所述非杂交区域包含选择对应于特定组蛋白修饰并且从而用作组蛋白修饰条形码的核酸序列,其中所述邻近探针的大小设置成同时允许所述组蛋白修饰结合结构域与所述组蛋白修饰结合并且所述互补核酸结合结构域与所述杂交核酸区域杂交;(c)在有效促进以下项的条件下用所述邻近探针孵育所述经修饰的无细胞核酸样品:(i)所述组蛋白修饰结合结构域与所述组蛋白修饰结合和(ii)所述互补核酸结合结构域与所述杂交核酸区域杂交以形成dsdna片段,所述dsdna片段具有源自所述无细胞dna的5'末端和源自所述邻近探针的3'末端;以及(d)通过添加聚合酶和dntp的混合物使所述片段的5'末端沿着所述邻近探针的非杂交区域和所述组蛋白修饰条形码延伸,从而提供组蛋白修饰条形码化的dsdna模板分子,用以进行进一步加工和测序;(e)将所述样品中的核酸纯化以提供包含组蛋白修饰条形码化的dsdna和dna的组合物;(f)从所述样品中的rna合成cdna的第一链;(g)合成cdna的与所述第一链互补的第二链以提供cdna双链体;以及(h)在不存在连接酶的情况下,将cdna衔接子共价附接至所述cdna双链体的至少一个末端,所述cdna衔接子包含含有源标识符条形码和rna指示剂条形码的序列,从而提供包含衔接子结合的cdna和所述组蛋白修饰条形码化的dsdna模板分子的核酸组合物。在该实施方案中,所述方法还包括(i)对组蛋白修饰条形码化的dsdna模板分子和所述衔接子结合的cdna进行扩增和测序,其中所述组蛋白修饰条形码化的dsdna模板分子和所述衔接子结合的cdna通常在合并的混合物中一起扩增和测序。在前述实施方案的一方面,所述方法还包括并入对在样品中的cfdna的分析以测定其羟甲基化图谱。所述方法包括进行实施方案的步骤(a)至(h),然后:(i)用第一亲和标签将所述核酸组合物中的5hmc残基官能化,所述第一亲和标签允许选择性去除含5hmc的物质;(j)从所述组合物中去除标签化的含5hmc的dna,无标签化的dna和衔接子结合的cdna保留;(k)将5hmc过程条形码附加到标签化的含5hmc的dna;(l)对所述含5hmc的dna、未标签化的dna(包括步骤(d)中产生的组蛋白修饰条形码化的dsdna模板分子)和衔接子结合的cdna进行扩增和测序,其中扩增和测序通常用各种物质的合并混合物进行。在实施方案的另一方面,所述方法还包括测定样品中cfdna的甲基化图谱。所述方法包括进行上述步骤(a)至(k),然后(1)将剩余样品中的甲基胞嘧啶残基转化为氧化的甲基胞嘧啶残基;(m)用第二种亲和标签将所述氧化的甲基胞嘧啶残基官能化,所述第二亲和标签允许从所述样品中选择性去除官能化的物质;(n)去除所述标签化的含5mc的dna,其中未标签化的dna和衔接子结合的cdna保留;(o)将5mc过程条形码附加到所述标签化的含5mc的dna;以及(p)对所述标签化的含5hmc的dna、所述标签化的含5mc的dna、所述未标签化的dna(包括如前所述的组蛋白修饰条形码化的dsdna模板分子)和所述衔接子结合的cdna进行扩增和测序,其中扩增和测序同样通常用各种物质的合并混合物进行。在本发明的另一个实施方案中,提供了一种组合工作流程方法,用于对血液样品进行血浆蛋白分析和对血液样品的无细胞核酸级分进行分析。血浆蛋白分析涉及使用邻近延伸测定法产生蛋白质条形码化的dsdna模板分子,并且最终将dsdna模板分子与一种或更多种在无细胞核酸样品分析中产生的各种dna模板分子(即组蛋白修饰条形码化的dsdna模板、标签化的含5hmc的dna、标签化的含5mc的dna、未标签化的dna和衔接子结合的cdna)合并。在另一个实施方案中,本发明提供了用于测定核酸模板分子的非经典序列特征的基于测序的方法,所述方法包括:将标识符序列附加到所述核酸模板分子,所述标识符序列指示所述模板分子的特定非序列特征;扩增所述核酸模板分子和附加的标识符序列,以得到多个扩增子,每个扩增子包括所述附加的标识符序列;以及对所述扩增子进行测序,并且从所获得的序列读取中测定所述非序列特征。本发明的另一个实施方案关于双链dna模板分子,其包含在邻近延伸测定法中衍生自已知蛋白质分析物的蛋白质特异性核酸序列,并且从而用作蛋白质标识符条形码。本发明的又另一个实施方案提供了样品级分的组合,每个样品级分包含源自单个血液样品的连接有衔接子的条形码化的双链dna模板分子,所述组合包括:(a)源自血浆的样品级分,其包含至少一种蛋白质相关的dsdna模板分子,每种dsdna模板分子包含对应于特定蛋白质分析物并且从而用作蛋白质标识符条形码的蛋白质特异性核酸序列;和(b)至少一个cfdna衍生的样品级分,其包含从所述血液样品中获得的无细胞核酸样品中获得的双链cfdna模板分子,其中所述cfdna模板分子与一组衔接子末端连接,所述衔接子包含选自源标识符条形码、片段标识符条形码、链标识符条形码、组蛋白修饰条形码、随机条形码及其组合的ufi序列。在本发明的又另一个实施方案中,上述样品级分的组合包含样品级分的合并混合物,其中然后可以同时对混合物中的dna模板分子进行扩增和测序。在本发明的另一个实施方案中,提供了方法和组合物,其用于提高衔接子连接效率,进而改进用于dna测序的方法。将理解的是,只要无细胞样品中dna的浓度已经非常低,本发明的上述方法和组合物在cfdna的分析中特别有用。此外,所述方法和组合物在含5mc的dna和含5hmc的dna的测序和定量中特别有用,因为这些经修饰的胞嘧啶残基相对不频繁出现,分别占所有dna碱基的约1%和0.1%。因此,任何用于检测5mc和5hmc,尤其是5hmc的方法都需要表现出相对于已鉴定的所有5hmc残基的级分的高效率以及高选择性,这意味着实际上基本上所有鉴定为5mc的残基应该是5mc残基,并且类似地,基本上所有鉴定为5hmc的残基应该是5hmc残基。然后,在另一个实施方案中,本发明提供了改进的方法和组合物,用于cfdna(例如,含有5mc残基、5hmc残基或5mc和5hmc残基二者的cfdna)测序,其中所述改进包括使用截短的测序衔接子,所述衔接子促进单个模板连接反应,使得仅在扩增(例如pcr扩增)时,样品才能对连接有衔接子的cfdna进行索引。在另一个实施方案中,提供了一种用于向dsdna分子添加标识符条形码的方法,其包括:(a)提供呈y-构建体形式的测序衔接子,其具有包含2个碱基对至50个碱基对的双链片段和两个各自包含2个碱基至25个碱基的单链片段;(b)将所述测序衔接子与末端钝化的a尾dsdna模板分子连接;(c)在pcr过程中使用至少一种条形码引物扩增所述连接有衔接子的dsdna模板分子,其中所述条形码引物包含:(i)第一区域,其不与所述衔接子中的任何序列互补并且包含标识符条形码;(ii)第二区域,其与所述衔接子的单链片段充分互补以与其杂交,使得在聚合酶的存在下所述条形码化引物的延伸产生所述引物的第二区域和所述衔接子的单链片段的双链复合物,其中包含所述标识符条形码的第一区域作为单链寡核苷酸尾部延伸超过所述双链复合物的末端。在一个相关实施方案中,本发明提供了一种用于扩增和测序dsdna模板分子的试剂盒,其包括:(a)呈y-构建体形式的测序衔接子,其具有包含2个碱基对至50个碱基对的双链片段和两个各自包含2个碱基至25个碱基的单链片段;(b)条形码化的引物,其包含(i)第一区域,其与所述衔接子中的任何序列不互补并且包含标识符条形码;(ii)第二区域,其与所述衔接子的单链片段充分互补以与其杂交;和(c)聚合酶。附图说明图1示意性地示出了使用邻近延伸测定法和蛋白质ufi序列将关于生物样品中的蛋白质分析物的信息转换成经典序列信息。图2示意性地示出了使用无细胞chip(cfchip)方法来使用邻近探针和组蛋白修饰ufi序列将与关于核小体中的组蛋白修饰的信息转换为经典序列信息。图3示意性地示出了本发明的全面的组合工作流程方法。图4示意性地示出了本发明的另一种组合工作流程方法,其中省略了cfrna分析。图5示意性地示出了本发明的另外的组合工作流程方法,其中不包括血浆蛋白质组学。图6示出了用于添加标识符序列(图中的“[索引]”)的现有技术模板/衔接子/引物构建体。图7示出了使用本发明的截短衔接子与条形码引物组合的相应构建体。图8示意性地示出了在pcr方法中索引引物和截短衔接子的使用。图9示出了如实施例2的部分(a)中所述的dna片段化后获得的大小分布图。图10是文库浓度(ng/μl)相对于衔接子输入浓度以及mt组全基因组测序(wgs)的图。图11以采样的模板的比例相对于假定的pcr效率的图示出了头对头衔接子的比较结果。图12提供了截短的衔接子效率和标准衔接子效率的并排比较,如实施例3所述。具体实施方式1.术语和概述:除非另有定义,否则本文所用的所有技术和科学术语具有本发明所属领域的普通技术人员通常理解的含义。以下定义对于本发明的描述特别重要的特定术语。其他相关术语在quake等“noninvasivediagnosticsbysequencing5-hydroxymethylatedcell-freedna.”的国际专利公开号wo2017/176630中定义。前述专利出版物以及本文所提及的所有其他专利文件和出版物明确地通过引用并入。在本说明书和所附的权利要求书中,除非上下文另外明确指出,否则单数名词包括其复数形式。因此,例如,“衔接子”不仅是指单个衔接子,而且是指可以相同或不同的两个或更多个衔接子,“模板分子”是指单个模板分子以及多个模板分子等。数字范围包括定义范围的数字。除非另有说明,否则分别地,核酸以5'至3'方向从左至右书写;氨基酸序列以氨基至羧基的方向从左至右书写。本文提供的标题不是对本发明的各个方面或实施方案的限制。相应地,下面整体上定义的术语通过参考整个说明书更完整地定义。除非另有定义,否则本文所用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的相同含义。singleton,等,dictionaryofmicrobiologyandmolecularbiology,第2版(newyork:johnwileyandsons,1994)和hale和markham,theharpercollinsdictionaryofbiology(newyork:harperperennial,1991)向本领域普通技术人员提供了本文所用的许多术语的一般含义。尽管如此,为了清楚起见和易于参考,下文定义了某些术语。如本文所用,术语“样品”涉及一种材料或材料的混合物,通常但不是必须的,呈液体形式,其包含一种或更多种目标分析物。如本文所用,术语“生物样品”涉及源自人受试者的生物流体、细胞、组织或器官的样品,其包含生物分子的混合物,该生物分子包括蛋白质、肽、脂质、核酸等。通常但不是必须的,样品是血液样品,例如全血液样品、血清样品或血浆样品。如本文所用的术语“核酸样品”是指包含核酸的生物样品。核酸样品可以是包含核小体的无细胞核酸样品,在这种情况下,核酸样品在本文中有时被称为“核小体样品”。核酸样品也可以由无细胞dna构成,其中样品基本上不含组蛋白和其他蛋白质,例如在无细胞dna纯化之后的情况。本文的核酸样品还可以包含无细胞rna。“样品级分”是指原始生物样品的子集,并且可以是生物样品在组成上相同的级分,如将血液样品分成相同的级分一样。可替代地,样品级分可以在组成上不同,例如当去除生物样品的某些组分时的情况,其中无细胞核酸的提取就是一个这样的实例。如本文所用,术语“无细胞核酸”涵盖无细胞dna和无细胞rna二者,其中无细胞dna和无细胞rna可以在包含体液的生物样品的无细胞级分中。体液可以是血液,包括全血、血清或血浆,或者可以是尿液、囊液或其他体液。在许多情况下,生物样品是血液样品,并且使用本领域普通技术人员已知的和/或在相关文本和文献中描述的现在常规的方法从其中提取无细胞核酸样品;用于进行无细胞核酸提取的试剂盒是可商购获得的(例如,dna/rnamini试剂盒和qiampdnabloodmini试剂盒,都可从qiagen获得,或magmaxcell-freetotalnucleicacid试剂盒和magmaxdnaisolation试剂盒,可从thermofisherscientific获得)。还参见例如hui等,fong等(2009)clin.chem.55(3):587-598。术语“核苷酸”旨在包括不仅包含已知的嘌呤和嘧啶碱基,而且还包含已被修饰的其他杂环碱基的那些部分。这样的修饰包括甲基化的嘌呤或嘧啶、酰化的嘌呤或嘧啶、烷基化的核糖或其他杂环。另外,术语“核苷酸”包括包含半抗原或荧光标记的那些部分,并且不仅可以含有常规的核糖和脱氧核糖,而且也可以含有其他糖。经修饰的核苷或核苷酸还包括对糖部分的修饰,例如其中一个或更多个羟基被卤素原子或脂肪族基团取代,或被官能化为醚、胺等。本文特别目标是经修饰的胞嘧啶残基,包括5-甲基胞嘧啶及其氧化形式,例如5-羟甲基胞嘧啶、5-甲酰基胞嘧啶和5-羧甲基胞嘧啶。术语“核酸”和“多核苷酸”在本文中可互换使用,以描述任何长度的聚合物,例如,由大于约2个碱基、大于约10个碱基、大于约100个碱基、大于约500个碱基、大于1000个碱基并且至多约10,000个或更多个碱基构成的核苷酸,例如脱氧核糖核苷酸或核糖核苷酸。核酸可以通过酶促法、化学合成或天然获得。如本文所用,术语“寡核苷酸”表示核苷酸的单链多聚体,其长度为约2至200个核苷酸、至多500个核苷酸。寡核苷酸可以是合成的或可以通过酶促法制备,并且在一些实施方案中,其长度为30至150个核苷酸。寡核苷酸可以包含核糖核苷酸单体(即可以是寡核糖核苷酸)和/或脱氧核糖核苷酸单体。寡核苷酸的长度例如可以为10至20、21至30、31至40、41至50、51至60、61至70、71至80、80至100、100至150或150至200个核苷酸。术语“杂交”是指核酸链通过本领域已知的碱基配对与互补链结合的过程。如果两个序列在中等至高度严格的杂交和洗涤条件下特异性地彼此杂交,则该核酸认为是与参考核酸序列“选择性杂交”。中等和高度严格的杂交条件是已知的(参见,例如ausubel等,shortprotocolsinmolecularbiology,第3版,wiley&sons1995和sambrook等,molecularcloning:alaboratorymanual,第3版,2001coldspringharbor,n.y.)。术语“双链体”和“双链体的”在本文中可互换使用,以描述碱基配对即杂交在一起的两个互补多核苷酸。dna双链体在本文中称为“双链dna”或“dsdna”,并且可以是完整分子或分子片段。例如,本文中称为条形码化和连接有衔接子的dsdna是完整分子,而在邻近延伸测定法中邻近探针的核酸尾部之间形成的dsdna是dsdna片段。如本文所用,术语“链”是指核酸的单链,其由通过共价键例如磷酸二酯键共价连接在一起的核苷酸制成。在细胞中,dna通常以双链形式存在,因此具有两条互补的核酸链,在本文中称为“顶”和“底”链。在某些情况下,染色体区域的互补链可以称为“加”和“减”链、“正”和“负”链、“第一”和“第二”链、“编码”和“非编码”链、“watson”和“crick”或“有义”和“反义”链。将链指示为顶链或底链是任意的,并且不意味着任何特定的方向、功能或结构。几个示例性哺乳动物染色体区域(例如bac、装配体,染色体等)的第一链的核苷酸序列是已知的,并且例如可以在ncbigenbank数据库中找到。术语“引物”是指合成的寡核苷酸,其在与多核苷酸模板形成双链体后,能够充当核酸合成的起始点,并且从其3'端沿模板延伸,因此形成延伸展的双链体。在延伸过程中添加的核苷酸的序列通过模板多核苷酸的序列测定。通常,引物通过dna聚合酶延伸。引物的长度通常与其在引物延伸产物的合成中使用的相容,并且长度通常为8至100个核苷酸,例如10至75、15至60、15至40、18至30、20至40、21至50、22至45、25至40等等。通常的引物的长度可以为10-50个核苷酸,例如15-45、18-40、20-30、21-25等等,并且在所述范围之间的任何长度。在一些实施方案中,引物通常不超过约10、12、15、20、21、22、23、24、25、26、27、28、29、30、35、40、45、50、55、60、65或70个核苷酸。如同用于引发dna复制反应的“引物”一样,“衔接子”也是短的合成寡核苷酸,其在生物学分析中具有特定目的。衔接子可以是单链或双链的,尽管本文优选的衔接子是双链的。在一个实施方案中,衔接子可以是发夹衔接子(即,一个与自身碱基配对以形成具有双链茎和环的结构的分子,其中该分子的3'和5'端连接至双链dna分子的5'和3'端)。在另一个实施方案中,衔接子可以是y-衔接子。在另一个实施方案中,衔接子本身可以由彼此碱基配对的两个不同的寡核苷酸分子构成。显而易见的是,衔接子的可连接端可以设计成与通过限制性酶切割产生的突出端相容,或者可以具有钝端或5't突出端。术语“衔接子”是指双链以及单链分子。衔接子可以是dna或rna,或两者的混合物。含有rna的衔接子可以通过rna酶处理或碱性水解来切割。衔接子可以为15至100个碱基,例如50至70个碱基,但是可以设想该范围之外的衔接子。如本文所用,术语“连接有衔接子的”是指已经与衔接子连接的核酸。衔接子可以连接至核酸分子的5'端和/或3'端。如本文所用,术语“添加衔接子序列”是指向样品中片段的末端添加衔接子序列的动作。这可以通过以下步骤完成:使用聚合酶填充片段的末端,添加a尾,并然后将包含t突出端的衔接子连接至a尾片段上。通常使用连接酶将衔接子连接至dna双链体,而对于rna,优选在不存在连接酶的情况下将衔接子共价或以其他方式连接至cdna双链体的至少一个末端。如本文所用,术语“不对称衔接子”是指衔接子,当连接至双链核酸片段的两端时,其将产生包含5'标签序列的顶链,该5'标签序列与3'端的标签序列不相同或互补。不对称衔接子的实例在weissman等的美国专利5,712,126和6,372,434以及bignell等的国际专利公开号wo2009/032167中进行了描述。可以通过两种引物扩增不对称标签化的片段:第一引物,其与添加至链的3'端的第一标签序列杂交;第二引物,其与添加至链的5'端的第二标签序列的互补序列杂交。y-衔接子和发夹衔接子(其可以在连接后被切割以产生“y-衔接子”)是不对称衔接子的实例。术语“y-衔接子”是指衔接子,其包含:双链区域和单链区域,其中相对序列不互补。双链区域的末端可以例如通过连接或转座酶催化的反应与靶分子(例如基因组dna的双链片段)连接。已经与连接有y-衔接子的衔接子标签化的双链dna的每条链是被不对称地标记的,因为它的一端具有y-衔接子的一条链序列,另一端具有y-衔接子的另一条链序列。两端已经与连接有y-衔接子的核酸分子的扩增产生不对称标签化的核酸,即,具有包含一个标签序列的5'端和有另一个标签序列的3'端的核酸。术语“发夹衔接子”是指呈发夹形式的衔接子。在一个实施方案中,在连接后,发夹环可以被切割以产生在末端具有非互补标签的链。在一些情况下,发夹衔接子的环可能包含尿嘧啶残基,并且可以使用尿嘧啶dna糖基化酶和核酸内切酶viii切割该环,尽管其他方法是已知的。如本文所用,术语“连接有衔接子的样品”是指已经与衔接子连接的样品。如通过以上定义所理解的,已经与不对称衔接子连接的样品包含在5'和3'端具有非互补序列的链。如本文所用,术语“扩增”是指产生模板核酸的一个或更多个拷贝或“扩增子”,例如可以使用任何合适的核酸扩增技术进行,例如以下技术,如pcr(聚合酶链反应)扩增(包括巢式pcr和多重pcr)、rca(滚动环扩增)、nasba(基于核酸序列的扩增)、tma(转录介导的扩增)和sda(链置换扩增)。应理解的是,扩增技术的组合可以在某些情况下有利地使用,例如,与rca组合的多重pcr(参见,例如,gong等(2018)rscadv.8:27375)、与定量pcr(qpcr)组合的rca(参见,例如liu等(2016)analsci.32:963-968)等。术语“富集(enrich和enrichment)”是指从不具有特征的分析物(例如,不包含羟甲基胞嘧啶的核酸)中部分纯化出具有某些特征的模板分子(例如,包含5-羟甲基胞嘧啶的核酸)。富集通常使具有特征的分析物的浓度相对于不具有特征的分析物增加至少2倍、至少5倍或至少10倍。富集后,样品中至少10%、至少20%、至少50%、至少80%或至少90%的分析物可以具有用于富集的特征。例如,富集的组合物中至少10%、至少20%、至少50%、至少80%或至少90%的核酸分子可以包含具有一个或更多个羟甲基胞嘧啶的链,该羟甲基胞嘧啶已经被修饰为包含捕获标签。如本文所用,术语“测序”是指获得多核苷酸的至少10个连续核苷酸的同一性(例如,至少20个、至少50个、至少100个或至少200个或更多个连续核苷酸的同一性)的方法。如本文所用,术语“二代测序”(ngs)或“高通量测序”是指illumina、lifetechnologies、roche等目前采用的所谓的“合成平行测序”或“连接测序”平台。二代测序方法还可以包括纳米孔测序方法(例如由oxfordnanoporetechnologies商业化的)、电子检测方法(例如由lifetechnologies商业化的iontorrent技术的)以及基于单分子荧光的方法(例如由pacificbiosciences商业化的)。如本文所用,术语“读取”是指测序系统的原始或加工输出,例如大规模并行测序。在一些实施方案中,读取本文描述的方法的输出。在一些实施方案中,可能需要修剪,过滤和对齐这些读取,产生原始读取、修剪读取、对齐读取。“ufi”是表征一组核酸分子的独特特征标识符。ufi可以是有时称为“条形码”(在本文中有时称为“ufi序列”或“ufi条形码”)的特定核酸序列,或它可以是由于糖基化、生物素化等而产生的化学标签。ufi也可以是不存在特定特征(例如附加或掺入的部分);例如,ufi可以是不存在特定条形码,或者不存在糖基化或生物素化的部分等。ufi序列通常是用于鉴定核酸分子的特征的相对较短的核酸序列。在本文中包含ufi的核酸模板分子及其扩增子有时被称为“条形码”模板分子或扩增子。ufi序列类型的实例包括但不限于以下:“分子ufi序列”(或“分子条形码”)是短的核酸序列,该核酸序列附加到样品中的每个核酸模板分子上,使得如果ufi序列具有足够的长度,则每个核酸模板分子附接至独特ufi序列。分子ufi序列通常设计为一串随机核苷酸,部分简并的核苷酸,或者在一些情况下,即在有限数量的模板分子中,限定核苷酸。如本领域中已知的,分子ufi序列可以用于解决和抵消扩增和测序仪错误,允许用户追踪复制并将它们从下游分析中去除,能够进行分子计数,并转而测定分析物浓度。参见,例如,casbon等(2011)nuc.acidsres.39(12):1-8。“样品ufi序列”(或“样品条形码”或“索引ufi”)是核酸序列,该核酸序列附加到样品中的每个核酸模板分子上,使得多个样品可以一起组合、加工和测序,其中样品ufi序列能够按样品对读取进行挑选和分组(即,多路分解)。样品ufi序列鉴定从中获得样品的个体。“源标识符序列”(或“源ufi”或“源条形码”)鉴定起始源。源ufi通常是样品ufi。然而,在某些情况下,例如,当从同一个体获得不同类型样品时(例如,血液样品、囊肿液等),源ufi将指示样品的生理来源,而不是从中获得样品的患者。当组合多个样品(包括从单个个体获得的两种或更多种样品类型)时,应使用样品条形码和源条形码二者。“片段标识符序列”(或“片段ufi”或“片段条形码”):在核酸样品(其中核酸包含许多片段的群体(如在无细胞dna中天然存在的,或可以是通过多种已知的片段化技术(例如,物理、超声、酶促等)基因工程化的)中,样品中的每个片段用相应的片段标识符序列条形码化。具有非重叠片段标识符序列的序列读取代表不同的原始核酸模板分子,然而具有相同片段标识符序列或基本上重叠的片段标识符序列的读取可能代表同一模板分子的片段,本文鉴定出的独特特征是片段所衍生自的模板核酸分子。“链标识符序列”(或“链ufi”或“链条形码”)独立地标记dna双链体的两条链中的每条链,从而可以测定读取所源自的链,即w链或c链。“蛋白质标识符序列”(或“蛋白质ufi”或“蛋白质条形码”)包含在杂交区域内,与其邻近或在其附近,该杂交区域是在与邻近探针特异性结合的相应蛋白质的存在下在邻近探针对的核酸尾部之间形成的。因此,当读取时,蛋白质标识符序列鉴定出由邻近探针对靶向的蛋白质分析物的存在。“组蛋白修饰标识符序列”(或“组蛋白修饰ufi”或“组蛋白修饰条形码”)用于本文所述的无细胞染色质免疫沉淀(cfchip)技术中来鉴定在核小体中鉴定的组蛋白修饰。组蛋白修饰标识符序列包含在杂交区域内,与其邻近或在其附近,该杂交区域是在探针的核酸尾部(即在探针的第一末端)与缠绕在阻挡周围的dna的末端之间形成的。探针的另一个末端与目标组蛋白修饰结合。因此,当读取时,组蛋白修饰标识符序列鉴定出组蛋白修饰的存在。“5hmc标识符序列”(或“5hmc条形码”)鉴定样品中的源自含5hmc的无细胞dna模板分子的dna片段,即“羟甲基化的”dna。“5mc标识符序列”(或“5mc条形码”)鉴定源自不包含5hmc的含5mc的无细胞dna模板分子的dna片段。“无细胞rna标识符序列”(或“cfrnaufi”)将cdna片段鉴定为源自cfrna模板分子。这些和其他ufi提供了将非经典序列特征——例如,血浆蛋白的存在和浓度、组蛋白修饰的位置和类型、羟甲基化图谱、甲基化图谱等——转化为可以从中得出非经典序列特征的经典序列数据的基础。本申请不限于前述类型的ufi,并且还可以设想其他类型的ufi。例如,基于非序列特征,可以使用许多类型的“过程标识符序列”或“过程ufi”来鉴定用于划分未扩增模板dna片段的初始库的许多过程中的任一个。除了组蛋白修饰ufi、蛋白ufi和表观遗传学ufi(包括5hmcufi和5mcufi)(其都可以被表征为过程ufi)之外,还有其他类型的可以有利地与本发明结合使用的ufi,包括表示模板分子序列之外的相邻基因组区域的存在或同一性的ufi,例如跨基因组跨度的ctcf结合位点。在一些实施方案中,ufi的长度可以为1至约35个核苷酸,例如2至30个核苷酸、4至30个核苷酸、4至24个核苷酸、4至16个核苷酸、4至12个核苷酸、6至20个核苷酸、6至16个核苷酸、6至12个核苷酸等。在某些情况下,如上所述,ufi可能进行错误检测和/或错误校正,意味着即使存在错误(例如,如果在导致测定分子条形码序列的各种加工步骤中的任一个过程中,分子条形码的序列合成错误、读错或失真),那么仍可以正确地解释代码。错误校正序列的使用在文献中进行了描述(例如,在hamati等的美国专利公开号us2010/0323348和braverman等的美国2009/0105959中,二者均通过引用并入本文)。可以使用任何有效方式将本文中用作ufi序列的寡核苷酸掺入dna分子中,其中“掺入”在本文中可与“添加到”和“附加到”互换使用,只要可以在dna分子的末端、dna分子的末端附近或dna分子内提供ufi即可。例如,可以使用选择的连接酶将多个ufi与dna末端连接,在这种情况下,只有最终ufi在分子的“末端”。另外,在下文详细描述的邻近延伸测定发和组蛋白修饰方法中,ufi可以包含在邻近探针的核酸尾部内、在邻近探针的核酸尾部末端或在探针与蛋白质靶标结合时产生的杂交区域内。如本文所用,术语“蛋白质分析物”涵盖多种肽物质,包括寡肽、多肽和蛋白质,其中,作为分析物的目标物质可以存在于或可以不存在于特定样品中。因此,在本文中样品中分析物的“检测”可以包括检测分析物的存在或不存在、确认分析物的可能存在、测定检测到的分析物的浓度等。更一般地,术语“检测”与术语“测定”、“测量”、“评价”、“评估”、“测定”和“分析”可互换使用,以指代任何形式的测量,并且包括测定元素是否存在。这些术语包括定量和/或定性测定。评估可以是相对的或绝对的。因此,“评估……的存在”包括测定存在部分的量,以及测定其是否存在。“羟甲基化水平”或“羟甲基化状态”是目标基因座内羟甲基化的程度。羟甲基化的程度通常以核酸区域内的羟甲基化密度来测量,例如,5hmc残基与经修饰和未经修饰的总胞嘧啶之比。羟甲基化密度的其他测量也是可能的,例如核酸区域中的5hmc残基与总核苷酸之比。“羟甲基化图谱”或“羟甲基化特征”是指包括在多个羟甲基化基因座的每一个处的羟甲基化水平的数据集。2.蛋白质分析物检测:在第一实施方案中,提供了一种用于检测生物样品中的蛋白质分析物的方法,并且该方法可以包括检测蛋白质分析物的存在以及定量,即测定分析物的量或浓度。与本文所述的其他方法一样,所寻求和获得的信息源自通过适当条形码化的核酸模板分子产生的序列读取。将理解的是,该方法扩展到对样品中多种蛋白质分析物的每一种的检测,如下所述。邻近连接测定法(pla)和邻近延伸测定法(pea)是用于检测和定量复杂生物样品中蛋白质的已建立方法。pla包括将蛋白质分析物与两个“邻近探针”结合,邻近探针是与dna链偶联的抗体。当链足够接近时(如探针与靶分析物结合时发生的情况),它们与dna连接酶联合。然后,连接产物用作定量pcr(qpcr)的模板,反映存在的蛋白质分析物的量。pea方法在蛋白质分析物的检测和定量中也是已知的,并且与pla相比有显著改进,因为pla导致值得注意的回收率损失,尤其是对于复杂的生物样品;参见,例如,lundberg等(2011)nuc.acids.res.39(15):1-8。像pla一样,pea依赖于两个邻近探针的使用,每个探针本质上是与dna链偶联的抗体。在pea中,探针对中一个探针的dna“尾”与该对的另一探针的dna尾杂交,导致在探针之间形成双链dna(dsdna)片段,该片段具有源自该对的第一邻近探针的5'端。然后采用聚合酶和dntp的混合物来使dsdna片段的5'末端沿着第二邻近探针延伸。如pla测定,然后邻近延伸产物在常规蛋白质测定法中用作qpcr的模板,以定量目标分析物。本发明消除了对定量pcr的需要,并且代替地使用测序,通常为ngs,以检测和定量生物样品中的至少一种蛋白质分析物。用本发明的改进的邻近延伸测定法产生的测序的核酸产物是经扩增的蛋白质条形码化的dsdna模板分子,即包含蛋白质特异性ufi序列的dsdna扩增子(或pcr产物)。对该蛋白质条形码化的扩增子进行测序,并对该序列读取进行反卷积,以从序列读取中观察到的蛋白质ufi序列测定蛋白质分析物的存在和数量。在一方面,本发明提供了一种改进的邻近延伸测定法,其通过提供多种探针对来鉴定生物样品中的多种蛋白质分析物,每种探针对包含第一邻近探针和第二邻近探针,其中每种探针对靶向特定蛋白质分析物,并且在相应的蛋白质分析物的存在下在每种探针对的探针之间产生双链dna(dsdna)片段,其中改进包括以下:将蛋白质特异性核酸序列掺入在探针对的探针之间产生的dsdna片段中,从而形成蛋白质条形码化的dsdna模板分子;对所述蛋白质条形码化的dsdna模板分子进行扩增和测序;和从产生的序列读取中观察到的蛋白质特异性ufi中鉴定生物样品中的蛋白质分析物。通常,通过将含ufi的衔接子与片段的至少一个末端末端连接,将蛋白质特异性ufi掺入dsdna片段中。该方法在图1中示意性地示出。最优地,在前述方法中产生的蛋白质条形码化的dsdna模板分子还设置有包含5hmc残基的捕获序列。捕获序列可以是单个5hmc残基,或它可以是包含单个5hmc残基的短寡核苷酸序列,或包含两个或更多个5hmc残基的短寡核苷酸序列序列。5hmc残基的存在允许通过用亲和标签(例如生物素)经5hmc官能化来捕获,从而能够用亲和素或链霉亲和素表面从样品或其级分中去除生物素化的物质。将理解的是,蛋白质条形码化的dsdna模板分子可以都具有相同的捕获序列,然而由不同探针对产生的每个dsdna模板分子具有对应于该探针对靶向的蛋白质分析物的独特蛋白质ufi序列。可以在单个寡核苷酸序列或衔接子中将蛋白质特异性ufi序列和捕获序列同时添加到探针之间产生的dsdna模板中。可替代地,可以首先添加蛋白质特异性ufi序列,然后添加捕获序列。为了追踪复制并能够分子计数以及偏移扩增和测序错误,还可以将分子ufi序列与蛋白质特异性ufi和任选的含5hmc的捕获序列一起附加到通过邻近延伸测定法产生的每个dsdna模板分子。为了测定生物样品中蛋白质分析物的浓度,上文所述的改进的邻近延伸测定法另外包括使用蛋白质浓度对照组合物。将指示特定蛋白质分析物的序列读取与由蛋白质浓度对照组合物产生的序列读取比较,该蛋白质浓度对照组合物在开始掺入生物样品中。蛋白质浓度对照组合物是本领域已知的,并且例如包括加标对照,其中在加工之前将已知浓度的蛋白质添加到样品中。在一些实施方案中,加标对照与浓度阶梯结合使用,其中对照组合物具有浓度范围内的不同浓度。本发明的其中从蛋白质条形码化的dsdna模板的序列读取中测定蛋白质分析物的方法的优点在于可以同时加工大量生物样品,例如血液样品或其级分,例如血浆样品或血清样品。可以容易地同时加工至少50个、至少100个、至少300个、至少500个、至少1000个或至少1500个或更多个生物样品。在可商购获得的微孔板的单个孔(例如96孔、384孔或1536孔板)中提供每个要加工的样品可以是方便的。另一个优点是能够对同一样品进行其他类型的分析,并且还可以通过测序获得其他信息,这将在下文进行详细解释。在上述实施方案的一种变型中,提供了一种相关的用于使用基于dna测序的技术鉴定生物样品中的多种蛋白质分析物的方法,其中该方法包括:(a)提供多种探针对,每种探针对靶向特定蛋白质分析物,并且包含在第一末端的蛋白质结合结构域、在相对的第二末端的核酸结合结构域和在其之间的非杂交核酸区域,其中(i)第一邻近探针和第二邻近探针的蛋白质结合结构域能够同时与同一蛋白质分析物上的不同结合位点结合,并且(ii)所述探针的核酸结合结构域彼此互补并且当所述第一邻近探针和所述第二邻近探针都与蛋白质结合并且足够邻近以发生杂交时杂交以形成dsdna片段;(b)在有效促进以下项的条件下用所述探针对孵育所述生物样品或其级分:(i)探针对中每个邻近探针的蛋白质结合结构域与相应的蛋白质分析物结合和(ii)核酸结合结构域彼此杂交以形成dsdna片段,所述dsdna片段具有源自所述第一邻近探针的5'末端和源自所述第二邻近探针的3'末端;(c)通过添加聚合酶和dntp的混合物将第一邻近探针的5'末端沿着第二邻近探针延伸,以在所述探针之间产生dsdna片段,所述dsdna片段掺入用作蛋白质标识符条形码的蛋白质特异性核酸序列和包含5hmc残基的捕获序列,其中(i)所述第一探针、所述第二探针或所述第一探针和所述第二探针二者的核酸结合区域包含所述捕获序列、所述蛋白质标识符条形码或所述捕获序列和所述蛋白质标识符条形码二者;(ii)所述dntp的混合物包含至少一种5hmc残基;和/或(iii)在聚合酶延伸后将衔接子连接至所述dsdna片段的末端,其中至少一个衔接子包含所述捕获序列、所述蛋白质标识符条形码或所述捕获序列和所述蛋白质标识符条形码二者,从而形成蛋白质-条形码化的dsdna模板分子,每个dsdna模板分子包含捕获序列;(d)对包含捕获序列的蛋白质条形码化的dsdna模板分子进行扩增和测序;以及(e)从步骤(b)中产生的序列读取中观察到的蛋白质标识符条形码中鉴定所述生物样品中的蛋白质分析物。本领域技术人员将理解,在所提及类型的邻近探针中,每个蛋白质结合结构域包含抗原,并且每个结合位点包含表位。还应注意,在前述方法的优选形式中,如前所述,生物样品通常是血液样品,并且对血液样品的级分进行蛋白质分析,例如样品的血清或血浆,通常是血浆。3.蛋白质测定法与无细胞样品分析的组合:在一个实施方案中,生物样品是血液样品,其中蛋白质分析物检测在样品的级分,通常是血浆级分上进行,并且如果期望的话,其他类型的分析在相同样品的无细胞级分上进行。这在图1中示意性地示出。在这种情况下,可以将经扩增的蛋白质条形码化的dsdna模板分子与通过分析无细胞样品级分产生的其他类型的经扩增的条形码化的dsdna模板分子一起在单个库中进行测序。这些衍生自无细胞样品级分加工的其他dsdna模板分子可以包括例如,具有组蛋白修饰ufi、5hmc相关的ufi、5mc相关的ufi、将dna双链体指示为衍生自无细胞rna的cdna的ufi等的模板分子。可以在扩增之前将各种类型的条形码化的dsdna模板分子合并,并在单次运行中一起扩增,或者可以在测序之前扩增条形码化的dsdna模板分子。当一种类型的模板分子比另一种模板分子要更大程度地扩增时,例如对于具有罕见特征并且因此以比其他模板分子低得多的浓度存在的模板分子,后种方法是有用的。例如,含5hmc的dsdna模板分子的浓度比其他模板分子显著更低。预扩增一组模板分子,然后在单次运行中合并和扩增所有模板分子,是适用于相同情况的另一种方法。预扩增包括从生物样品或模板分子的混合物中分离出具有条形码化特征的一组模板分子,然后与剩余的模板分子重组并同时扩增。组合前述部分中所述的邻近延伸方法,从包含蛋白质分析物的相同生物样品中提取的无细胞核酸样品获得的信息可以包括检测无细胞核酸样品中的核小体内一种或更多种组蛋白修饰的存在、同一性、位置或数量(或其组合)。组蛋白修饰包括翻译后修饰(ptm),其中许多已被建立以通过改变染色质结构或以其他方式来调整基因表达。本文特别的目标组蛋白修饰是包含用于评估受试者的疾病状态的组蛋白修饰生物标志物的那些。根据本发明的该实施方案和其他实施方案的用于检测组蛋白修饰的方法在下一部分中进行了描述。从无细胞样品获得的其他信息可以包括:至少一种无细胞dna序列;至少一种无细胞rna序列;dna甲基化数据;dna羟甲基化数据;及可能与前述任何内容都不相关的其他信息。下文包括有关用于获得前述信息的适当和优选方法的详细信息。4.使用无细胞chip(cfchip)检测组蛋白修饰:在本发明的另一个实施方案中,提供了以下方法,其用于(1)制备无细胞核酸样品以能够使用基于测序的技术鉴定其中所含核小体中的至少一个组蛋白修饰,和(2)在含有完整核小体的无细胞核酸样品中检测组蛋白修饰,其中检测组蛋白修饰的存在、同一性、位置或数量,或其组合。两种方法都包括分析从生物样品(例如血液样品)中提取的无细胞核酸样品,其中无细胞核酸样品包含完整的核小体。核小体是染色质结构的基本单位,并且由八个高度保守的核心组蛋白的蛋白质复合物(其中每个核心组蛋白h2a、h2b、h3和h4的两个拷贝)构成。dna的约146个碱基对缠绕在组蛋白八聚体周围,以形成核小体“核心”。核心颗粒通过长度为至多约80个碱基对的接头dna延伸连接,其看起来像“绳珠”(koller等(1979)j.cellbiol.83(2pt1):403-427),直到用接头组蛋白(例如h1、h5或其同工型)压实以形成染色质。还已知核小体位置和核小体结构(就组成组蛋白蛋白质变体和翻译后修饰或ptm而言)介导表观遗传学信号传导。组蛋白ptm已经与多种过程联系,包括转录、dna复制和dna损伤。ptm通常位于核心组蛋白的尾部上,并且包括乙酰化、甲基化、二甲基化、三甲基化、丙酰化、丁酰化、巴豆酰化、2-羟基-异丁酰化、丙二酰化、琥珀酰化、甲酰化、泛素化、瓜氨酸化、磷酸化、羟基化、类泛素化、o-glcnac糖基化和adp核糖基化,并且更常见的修饰包括赖氨酸残基的乙酰化、甲基化或泛素化以及精氨酸残基的甲基化和丝氨酸残基的磷酸化。其功能已经相当好建立的ptm的全面列表在赵(2015),"appendix2-comprehensivecatalogofcurrentlydocumentedhistonemodifications,"coldspringharborperspectivesinbiology.2015;7(9):a025064中列出,其公开内容通过引用并入本文;还参见可从reactionbiologycorp.获得的histonemodificationposter,其提供了组蛋白修饰图以供参考[2018年8月5日在www.reactionbiology.com/webapps/site/检索]。已经通过elisa检测了单核小体和寡核小体,如salgame等(1997)nuc.acids.res.25(3):680-1和vannieuwenhuijze等(2003)ann.rheum.dis.62(1):10-14中报告的。此类测定法通常采用抗组蛋白抗体(例如抗h2b、抗h3或抗h1、h2a、h2b、h3和h4)作为捕获抗体,并且采用抗dna或抗h2a-h2b-dna复合抗体作为检测抗体。然而,这些方法和已经开发的其他方法往往具有有限的可靠性。本发明的方法“无细胞染色质免疫沉淀”(cfchip)测定法与现在常规的chip方法不同,其通过交联活细胞或组织中染色质中的dna和相关蛋白来分析蛋白质与dna之间的细胞内相互作用,通过超声处理或消化来剪切交联的复合物,免疫沉淀所得的交联的蛋白质-dna片段,对片段进行纯化和测序,并然后从序列读取中测定关于细胞中的蛋白质与dna之间的相互作用的信息。考虑到不同应用已经开发了该基本程序的多种变型,包括天然chip(nchip)、无磁珠chip、载体chip(cchip)、快速chip(qchip)、快速定量chip(q2chip)、microchip(μchip)、基质chip、病理-chip(pat-chip)等。在一个实施方案中,本发明提供了一种用于制备无细胞核酸样品以能够使用基于dna测序的技术鉴定其中所含核小体中至少一个组蛋白修饰的方法,其中该方法包括以包含多个核小体的无细胞核酸样品开始,每个核小体包含缠绕在组蛋白核心周围(即,由四种核心组蛋白中每一种的一对构成的组蛋白八聚体周围)的cfdna分子。将包含末端杂交区域的衔接子(例如,y-衔接子)连接至每个组蛋白相关的cfdna分子的末端。如本文前面所解释的,衔接子可以包含样品ufi序列和分子ufi序列。衔接子的连接在图2的顶部示出,并且产生经修饰的无细胞核酸样品,该样品包含各自缠绕在组蛋白核心周围的连接有衔接子的cfdna分子。在该方法的下一步骤中,也如图1中所示,采用邻近探针,该邻近探针包含在第一末端的特异性结合目标组蛋白修饰的组蛋白修饰结合结构域;在第二末端的与由衔接子提供的末端杂交区域之一互补的核酸结合结构域。组蛋白修饰结合结构域与核酸结合结构域之间的非杂交区域包含选择对应于目标组蛋白修饰的核酸序列,并且该核酸序列用作组蛋白修饰ufi(或组蛋白修饰“条形码”)。邻近探针的大小设置为以同时允许探针的组蛋白修饰结合结构域与目标组蛋白修饰结合和探针的互补核酸结合结构域与杂交核酸区域杂交。为了实现同时结合,在有效促进以下项的条件下将经修饰的无细胞核酸样品与邻近探针一起孵育:(i)探针的组蛋白修饰结合结构域与组蛋白修饰的结合和(ii)探针的互补核酸结合结构域与杂交核酸区域的杂交。这导致形成dsdna片段,其具有源自无细胞dna的5'末端和源自邻近探针的3'末端并且包含组蛋白修饰条形码。在dsdna片段形成之后,通过添加聚合酶和dntp的混合物以与该部分的第(2)部分中关于邻近延伸测定法所述的类似的方式使dsdna片段的5'末端沿着邻近探针的非杂交区域和组蛋白修饰ufi延伸。聚合酶延伸提供了组蛋白修饰条形码化的dsdna模板分子,通常也经如上所示的样品ufi序列和分子ufi序列进行条形码化,其然后可以经受扩增和测序。通常,上述cfchip方法包括使用多种探针对,每种探针对靶向不同组蛋白修饰,从而可以对在最后步骤中获得的序列读取进行反卷积以推导关于多种组蛋白修饰的信息,例如组蛋白ptm。在另一个相关实施方案中,提供了一种基于测序的方法,用于检测包含完整核小体的无细胞核酸样品中的组蛋白修饰,其中检测组蛋白修饰的存在、同一性、位置或数量,或其组合。该方法包括进行上述用于制备无细胞核酸样品以能够使用基于dna测序的技术鉴定至少一种组蛋白修饰的方法,然后扩增组蛋白修饰条形码化的dsdna模板分子,对所得扩增子进行测序,并从序列读取中观察到的组蛋白修饰ufi测定关于组蛋白修饰的类型和位置的信息。5.具有含5hmc的捕获序列的衔接子:在数个实施方案中,提及衔接子的使用,该衔接子包含含有至少一个5hmc残基的捕获序列。这是本文许多实施方案的任选特征,并且在组合工作流程方法中特别有用,在组合工作流程方法中,将连接有特异性衔接子的dsdna模板分子从样品或dsdna模板分子的混合物中下拉出,然后剩余组分在不存在去除的dsdna模板的情况下进行加工。下拉的模板分子可以单独扩增或只是留置,而样品的剩余物经受化学加工,最终将从单个样品产生的所有dsdna模板分子合并并一起测序。通过衔接子掺入dsdna模板分子中的捕获序列有助于从样品中下拉或去除dsdna模板分子。捕获序列包含5hmc残基;该序列可以是单个5hmc残基、包含单个5hmc残基的短核酸序列或包含两个或更多个5hmc残基的短核酸序列。在一个实施方案中,将包含含5hmc的捕获序列的衔接子连接至dsdna模板分子的至少一个末端,或在cfrna分析中,通过无连接酶的化学方法附接至cdna分子的至少一个末端。当要下拉衔接子结合的dsdna模板分子时,用亲和标签将衔接子中的5hmc残基官能化,该亲和标签允许选择性去除亲和标签化的模板。在一个实施方案中,亲和标签包括生物素部分,例如生物素、脱硫生物素、氧生物素、2-亚氨基生物素、二氨基生物素、生物素亚砜、生物胞素等。使用生物素部分作为亲和标签允许方便地用亲和素或链霉亲和素表面(例如链霉亲和素珠、磁性链霉亲和素珠等)去除。用生物素部分或其他亲和标签标记5hmc残基通过将化学选择性基团共价附接至衔接子中的5hmc残基上来完成,其中化学选择性基团能够与官能化的亲和标签进行反应从而将亲和标签连接至5hmc残基。在一个实施方案中,化学选择性基团是udp葡萄糖6-叠氮化物,其与炔烃官能化的生物素部分进行自发的1,3-环加成反应,如robertson等(2011)biochem.biophys.res.comm.411(1):40-3、he等的美国专利号8,741,567和quake等的wo2017/176630中所述的。因此,炔烃官能化的生物素部分的添加导致生物素部分共价附接至每个5hmc残基。这种反应的实例在he等的美国专利号8,741,567的图5b中示出,其通过引用并入本文。然后如上所示,可以使用亲和素或链霉亲和素表面将亲和标签化的dsdna模板分子下拉,并留置用于以后加工和分析。去除亲和标签化的片段后剩余的上清液包含dsdna模板分子,该dsdna模板分子的内部序列或所附接的衔接子中不包含5hmc。剩余的dsdna模板分子可以继续进行化学加工,并最终与下拉的模板分子重合用以进行测序。本发明涵盖作为新的物质组合物的含5hmc的衔接子结合的cfdna模板分子,其中所述衔接子可以包含除至少一个5hmc残基之外的ufi序列,例如源ufi序列、分子ufi序列、链标识符ufi序列或组蛋白修饰ufi序列,如本文前面所解释的。6.无细胞rna的分析:无细胞rna,主要源自凋亡小体和外泌体,通常高度降解,具有非常短的半衰期,并且以非常低的浓度存在于无细胞样品中。因此,从cfrna制备cdna测序文库具有挑战性,因为cfrna的低完整性消除了在制备cdna文库中使用标准rna-seq方法的可能性。可以适用于本文的方法是采用无连接的cdna合成和文库制备技术的那些,其中扩增所需的衔接子共价附接至cdna而无需连接酶。在一种这样的方法中,使用随机引物来从cfrna,优选从贫rrna的rna合成cdna,如可以用rna酶制备的;参见sooknanan的美国专利号9,745,570,其公开内容通过引用并入本文。可以使用任何有效的末端标签化程序引入用于进行扩增和条形码化的5'和3'接头标签(即,将cfrnaufi序列添加至cdna),例如在sooknanan的美国专利号8,304,183中描述的方法,也通过引用并入。该方法可以使用可商购获得的试剂盒进行,例如可从epicenterbiotechnologies(illumina,inc.)获得的scriptseqtmv2rna-seq文库制备试剂盒。与scriptseqcdna文库制备结合使用的材料、试剂和方法的其他说明可以在scriptseqtmv2rna-seq文库制备指南中找到[在2018年8月16日从support.illumina.com检索的]。在不使用衔接子连接的情况下从cfrna制备cdna文库的另一种方法中,用包含衔接子序列的dt引物的3'-聚腺苷酸化rna的第一链cdna合成采用了模板转换技术,该技术利用所选逆转录酶的末端转移酶活性。当到达rna的5'端时,非模板核苷酸的短序列(例如ccc)延伸cdna的第一链,并且包含与所添加序列互补的短序列(例如ggg)的模板转换寡核苷酸和用作正向pcr引物的第二衔接子序列与第一链延伸杂交,并能够通过pcr合成和扩增第二链。参见betts等的美国专利公开号2017/0198285a1;zhu等(2001)biotechniques30(4):892-897;和"technote:asmarterapproachtosmallrnasequencing,",网络文件重印[在2018年8月16日从https://www.takarabio.com/learning-centers/next-generation-sequencing/technical-notes/full-length-small-rna-libraries检索],其公开内容通过引用整体并入本文。在本发明的上下文中,使用无连接酶的方法(例如上述技术之一)来从生物样品中的cfrna合成衔接子结合的cdna,其中该衔接子包含cfrnaufi序列,以将dsdna模板分子鉴定为cfrna衍生的cdna。衔接子还包含至少一种另外的ufi序列,例如源ufi序列、分子ufi序列、链标识符ufi序列或组蛋白修饰ufi序列,如本文前面所解释的。然后可以对衔接子结合的cdna进行扩增和测序,并且可以通过对序列读取进行反卷积获得有关生物样品中cfrna的信息。cfrna可以是不翻译成蛋白质的mrna或rna,即非编码rna(ncrna),例如trna;rrna;小rna,例如microrna(mirna)、sirna、pirna、snorna、snrna、exrna和scarna;以及长nrna,例如xist和hotair。该部分中前面所总结的scriptseq方法最适于与已去除rrna的无细胞rna样品结合使用,而clontechsmart方法与小ncrna结合特别有用。此时可以对结合衔接子的cdna进行扩增和测序,或者可以在扩展的组合工作流程方法的背景下进行进一步分析。如第8部分所解释的,特别感兴趣的是dsdna的羟甲基化和/或甲基化分析。7.组合工作流程中无细胞rna和无细胞dna的分析:在一个实施方案中,提供了一种在单个无细胞核酸样品中制备dna和rna用以同时进行基于测序的鉴定的组合工作流程方法。在从生物样品中提取无细胞核酸样品后,此处初始步骤是将选择的衔接子连接至cfdna。可以将衔接子连接至无细胞核酸样品中cfdna片段的末端,以形成连接有衔接子的dsdna模板分子。可以使用标准的连接条件和可商购获得的连接酶。选择用于与cfdna片段连接的衔接子包含样品ufi序列和优选地至少一种另外的ufi序列,例如分子ufi序列和链标识符ufi序列。然后使用常规的核酸纯化技术将连接有衔接子的cfdna与cfrna一起纯化,以提供cfrna和连接有衔接子的dna模板分子的无细胞混合物。在组合工作流程的下一步骤中,在仍然包含连接有衔接子的cfdna的无细胞混合物中加工cfrna,因为本发明的方法避免了在cdna合成之前或期间去除连接有衔接子的cfdna的需要。从cfrna合成cdna的第一链,然后合成cdna的与第一链互补的第二链,如本领域已知的,以形成cdna双链体。如前述部分所述的进行cdna合成,从而在无需连接酶的情况下将衔接子附接至cdna。如前所述,cdna衔接子包含源标识符ufi和rna指示剂ufi,从而在无细胞混合物中提供衔接子结合的cdna,该无细胞混合物还包含连接有衔接子的dna。此时可以对连接衔接子的dsdna模板分子和cdna模板分子进行扩增和测序,或者可以在扩展的组合工作流程方法的背景下进行进一步分析,包括dsdna的羟甲基化和/或甲基化分析,如以下部分所解释的。8.与羟甲基化和/或甲基化分析组合的工作流程:细胞中基因表达的表观遗传学控制部分地通过对dna核苷酸的修饰来介导,该修饰包括dna的胞嘧啶甲基化状态和胞嘧啶羟甲基化状态。在一段时间内本领域已知dna可以在胞嘧啶核苷酸的5位被甲基化以形成5-甲基胞嘧啶。报道了5-甲基胞嘧啶形式的甲基化dna发生在dna序列中胞嘧啶核苷酸紧邻鸟嘌呤核苷酸的位置。这些位置简称为“cpg”,并且基因组中包含高比例cpg位点的区域通常称为“cpg岛”;大多数人基因启动子序列与这样的cpg岛有关。在活性基因中,这些cpg岛通常被低甲基化。基因启动子序列的甲基化与稳定的基因失活有关。在癌细胞中观察到的dna甲基化模式不同于健康细胞的那些。报道了相对于健康细胞,重复元件,特别是围绕着着丝粒的区域,在癌症中被低甲基化,但是已经报道,特定基因的启动子在癌症中被高甲基化。据报道,这两种作用的平衡导致癌细胞中的整体dna低甲基化。已经使用免疫组织化学(ihc)技术以及许多其他方法研究了细胞中的整体dna甲基化,但是这些方法中的许多是不利的,因为它们劳动强度大和/或需要大量高质量的提取dna。当前用于检测整体dna甲基化的方法包括dna的提取或纯化,并且不适用于快速、高通量、低成本的微创诊断方法。类似地,只能通过分析基本上纯的或提取的dna来研究dna的其他经修饰或异常的碱基(例如尿嘧啶、肌苷、黄嘌呤和次黄嘌呤)的分析。不可能在复杂的生物介质(例如组织裂解液、血液、血浆或血清)中直接进行这样的分析。随着表观遗传学领域的发展,另一种dna修饰(在5位羟甲基化的胞嘧啶(5hmc))的检测已经被证明与5mc的检测同样重要。尽管5mc修饰通常发生在cpg二核苷酸内,但是天然5hmc残基倾向于出现在其他位置。5hmc的发生频率比5mc的发生频率低得多,比率通常为大约10:1,取决于组织类型(参见nestor等(2012)genomebiology13:r84),其中5mc占所有dna碱基的约1%和5hmc占所有碱基的约0.01%。尽管已经建立5hmc参与多种过程,包括转录、dna去甲基化,以及在异常5hmc模式的情况下参与肿瘤发生,但是5hmc的分子功能才开始被理解。参见tahiliani等(2009)science324(5929):930-035(2009);guo等(2011)cell145:423-434;wu等(2011)genes&development25:679-684;ko等(2010)nature468:839-843;和robertson等(2011)biochem.biophys.res.comm.411(1):40-3。还已知5hmc是稳定的dna修饰,通过10-11易位(tet)酶(例如tet1)将5mc催化氧化而形成的。亚硫酸氢盐测序不区分5mc与5hmc,并且因此,用于单独检测5mc和5hmc残基的其他方法是必须的。如上所示,5hmc看起来远低于5mc,因此关于已鉴定的所有5hmc残基的分数以及高选择性,用于检测5hmc的任何方法需要表现出高效率,这意味着基本上所有已鉴定为5hmc的残基实际上应该是5hmc残基。已经报道了数种用于检测dna中的5hmc的方法,这些方法包括用t4噬菌体酶、β-葡萄糖基转移酶(β-gt)进行糖基化,因为该酶选择性地将5hmc糖基化而不修饰5mc,在本发明的上下文中,组合工作流程方法优选地包括用于检测无细胞dna中经修饰的胞嘧啶残基(即5mc、5hmc或5mc和5hmc二者)的基于测序的方法。如果将羟甲基化分析与甲基化分析一起进行,则羟甲基化应该是最初的重点,然后是甲基化,如将从以下工艺流程描述中理解的。假设已经从生物样品中提取了无细胞核酸样品,其中已经将cfdna进行衔接子连接,然后进行cfrna加工以提供衔接子结合的cdna(如第7部分所述),则组合工作流程方法继续以提供关于cfdna的羟甲基化图谱的信息。“羟甲基化图谱”可以是羟甲基化密度,例如,核酸区域内5hmc残基与经修饰和未经修饰的总胞嘧啶之比。还设想了5hmc密度的其他测量,例如,基因座中5hmc残基与总核苷酸之比。除了5hmc密度之外,羟甲基化图谱还可以包括羟甲基化信息,例如羟甲基化模式、核酸区域内的总5hmc残基、核酸区域内5hmc残基的位置、核酸区域内5hmc残基的相对位置和/或羟甲基化位点为半羟甲基化或完全羟甲基化的鉴定。一种用于检测核酸的羟甲基化图谱的优选方法在quake等的国际专利公开wo2017/176630中进行了描述,其通过引用整体并入本文。该方法关于在测序方案的背景下检测无细胞dna中的5-羟甲基胞嘧啶模式。将亲和标签附加到无细胞dna样品中的5hmc残基上,并然后将标签化的dna分子富集和测序,其中鉴定5hmc位置。如quake等所述,该方法的一个示例性实例包括最初修饰无细胞样品中的末端钝化的连接有衔接子的双链dna片段,以将作为亲和标签的生物素共价附接至5hmc残基。这可以通过在6位用叠氮化物部分官能化的尿苷二磷酸(udp)葡萄糖选择性地将5hmc残基葡萄糖基化来进行,该步骤之后,如先前在第5部分中关于衔接子中含5hmc的捕获序列所述的,通过“点击化学”反应用炔烃官能化的生物素进行1,3-环加成反应。包含生物素化的5hmc残基的dna片段是连接有衔接子的dsdna模板分子,然后可以在“富集”步骤中将其用链霉亲和素珠下拉。在本发明的组合工作流程方法中,将5hmcufi序列添加到下拉的连接有衔接子的dsdna模板分子的末端,因此在扩增、合并和测序后,可以从获得的序列读取推断关于羟甲基化图谱的信息。即,分析序列读取以定量测定在cfdna中哪些序列被羟甲基化。这可以通过例如计数序列读取或可替代地,在扩增之前对原始起始分子的数目进行计数来完成,基于其断裂点和/或它们是否包含相同的分子ufi。用于测定无细胞核酸样品中dna的羟甲基化图谱的其他方法在arensdorf等的于2018年2月14日提交的临时美国专利申请序列号62/630,798的“methodsfortheepigeneticanalysisofdna,particularlycell-freedna,”和给song等的美国专利公开号2017/0298422中进行了描述,其二者均通过引用并入本文。这些参考文献也可与本发明的实施方案结合使用,其中本发明的组合工作流程方法还包括检测除cfdna羟甲基化图谱之外的cfdna甲基化谱。在本发明的组合工作流程方法的上下文中,可以如下执行arensdorf方法:双生物素技术:在已经从生物样品中提取无细胞核酸样品,其中将cfdna与衔接子连接,然后进行cfrna加工以提供衔接子结合的cdna(如第7部分所述)之后,cfdna中的5hmc残基被亲和标签(例如生物素部分)选择性地标记,,如本文前面所解释的。可以通过以下进行生物素化:通过βgt催化的葡萄糖基化用尿苷二磷酸葡萄糖-6-叠氮化物将5hmc残基选择性地官能化,然后进行点击化学反应以共价附接炔基官能化的生物素部分,如前面所解释的。然后使用亲和素或链霉亲和素表面(例如,以链霉亲和素珠的形式)拉出在5hmc位置生物素化的所有dsdna模板分子,然后将其置于单独的容器中用以在扩增过程中进行ufi序列附接。上清液中剩余的dsdna模板分子是具有5mc残基或无修饰的片段(后组包括从cfrna产生的cdna)。然后使用tet蛋白将上清液中的5mc残基氧化为5hmc;在这种情况下,采用tet突变蛋白来确保5mc的氧化不会超过羟基化。出于该目的,合适的tet突变蛋白在liu等(2017)naturechem.bio.13:181-191中进行了描述,其通过引用并入本文。然后重复βgt催化的葡萄糖基化,然后进行生物素官能化。如此标记的片段-在每个原始5mc位置生物素化的-用链霉亲和素珠下拉。然后,在扩增过程中,将珠结合的dna片段条形码化-用第一步骤中使用的ufi序列,即5mcufi序列。未经修饰的dna片段,即不含经修饰的胞嘧啶残基的片段,现在保留在上清液中。如果期望的话,可以使用序列特异性探针与未甲基化的dna链杂交。如前所述,可以将得到的杂交复合物拉出,并在扩增过程中用另外的ufi序列标签化。pic-硼烷方法:这是双重生物素技术的替代,并且还从连接有衔接子的dna片段中5hmc残基的生物素化开始,然后亲和素或链霉亲和素下拉。然而,在该技术中,上清液中保留的包含未经修饰的5mc残基的dna被氧化超过5hmc,变成5cac和/或5fc残基。氧化可以使用催化活性的tet家族酶以酶促方式进行。如本文所用的那些术语“tet家族酶”或“tet酶”是指如在美国专利号9,115,386中所定义的催化活性的“tet家族蛋白”或“tet催化活性片段”,其公开内容通过引用并入本文。在该上下文中,优选的tet酶是tet2;参见ito等(2011)science333(6047):1300-1303。氧化也可以使用化学氧化剂化学地进行。合适的氧化剂的实例包括但不限于:无机或有机过钌酸盐的形式的过钌酸盐阴离子,包括金属过钌酸盐如过钌酸钾(kruo4)、四烷基铵过钌酸盐如四丙基过钌酸铵(tpap)和四丁基过铵钌(tbap)、以及聚合物支撑的过钌酸盐(psp);无机过氧化合物和组合物,例如过氧钨酸盐或高氯酸铜(ii)/tempo组合。此时,不必将含5fc的片段与含5cac的片段分离,只要在该方法的下一步骤中,将5fc残基和5cac残基二者转化为二氢尿嘧啶(dhu)。即,在将5mc残基氧化为5fc和5cac之后,添加有机硼烷以使氧化的5mc残基还原、脱氨基和脱羧或去甲酰化。所得的dsdna模板分子包含dhu代替原始的5mc残基,并且可以与源自同一样品的其他dsdna模板分子一起进行扩增、合并和测序。有机硼烷可以表征为硼烷和选自氮杂环和叔胺的含氮化合物的复合物。氮杂环可以是单环、双环或多环的,但是通常是单环的,其形式为5或6元环,其含有氮杂原子和任选地选自n、o和s的的一个或更多个另外的杂原子。氮杂环可以是芳香族或脂环族的。在本文中优选的氮杂环包括2-吡咯啉、2h-吡咯、1h-吡咯、吡唑烷、咪唑烷、2-吡唑啉、2-咪唑啉、吡唑、咪唑、1,2,4-三唑、1,2,4-三唑、哒嗪、嘧啶、吡嗪、1,2,4-三嗪和1,3,5-三嗪,其中的任一种可以未被取代或被一个或更多个非氢取代基取代。典型的非氢取代基是烷基,特别是低级烷基,如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基等。示例性的化合物包括吡啶硼烷、2-甲基吡啶硼烷(也称为2-甲基吡啶硼烷)和5-乙基-2-吡啶。关于这些有机硼烷及其将氧化的5mc残基转化为dhu的反应的进一步信息可以在以上引用的arensdorf专利申请、临时美国专利申请序列号62/630,798中找到,其前面通过引用并入本文。生物素/天然5mc富集方法:这是双重生物素技术的替代,并且从连接有衔接子的dna片段中5hmc残基生物素化开始,然后亲和素或链霉亲和素下拉。然而,在此,替代修饰上清液中保留的甲基化dna,使用抗5mc抗体或mbd蛋白捕获并下拉天然含5mc的片段。该技术在本文中较不优选,因为它不会产生可以与衍生自同一样品的其他dsdna模板分子进行扩增、合并和测序的dsdna模板分子。因此,产生的条形码化的连接有衔接子的dsdna模板分子是含有5hmc的dsdna和含有5mc且不含5hmc的dsdna,并且任选地还包含不含经修饰的胞嘧啶残基的dsdna。这些模板分子与以下中的至少一种一起扩增、合并和测序:通过第2部分的方法产生的组蛋白修饰条形码化的dsdna模板分子;如第6部分所述,来自cfrna加工的衔接子结合的cdna;和通过第2部分中所述的方法从相同生物样品(例如血液样品)产生的蛋白质条形码化的dsdna模板分子。因此,在单次运行中对上述混合物进行测序可以提供关于核小体,特别是组蛋白修饰的信息;cfrna序列;蛋白质分析物身份和浓度;cfdna羟甲基化图谱;和cfdna甲基化图谱。单链cfdna:以上和本文其他地方所述的方法也可以适于在单链cfdna的分析中进行,例如以测定甲基化图谱、羟甲基化图谱等。在本领域技术人员已知的和/或在相关文本和文献中所述的rca技术的背景下,可以使用rca引物将选择的ufi序列(例如分子ufi序列、样品ufi序列、过程ufi序列(包括5hmcufi序列和5mcufi序列,如上所解释))引入单链cfdna中。可替代地,对于单链cfdna,可以产生互补链,并然后对dsdna分子进行本发明的方法,如本文其他地方所述。9.全面的组合工作流程方法:因此,本发明提供了一种组合工作流程方法,其中通过在单次运行中将被标签化以指示生物样品的各种特征的dsdna模板分子的扩增子进行合并和测序,从单个生物样品中获得多种类型的信息。该方法的最全面版本在图3中示意性地示出并且包括以下步骤:(1)如第2部分中所述的,使用邻近延伸测定法从受试者获得的包含多种蛋白质分析物的生物样品(例如血液样品)级分中产生蛋白质条形码化的dsdna模板分子,其中每个具有特定蛋白质ufi序列的dsdna模板分子对应于样品中该特定蛋白质的存在;(2)以使其中的核小体保持完整状态的方式从生物样品的剩余物中提取无细胞核酸样品;(3)将衔接子连接至核酸样品中的无细胞dna,其中衔接子包含用于鉴定dna的来源或样品的源标识符ufi、用于将每个cfdna片段鉴定为样品中的原始分子的“随机”分子标识符ufi以及任选地用于将每个cfdna片段的链鉴定为c或w的“链”标识符ufi;(4)在不分离或分离核酸样品的任何组分的情况下,进行第4部分中所述的邻近延伸cfchip方法,以产生与使用邻近探针鉴定的组蛋白修饰相对应的dsdna模板分子,如此提供的dsdna模板分子具有对应于特定组蛋白修饰的组蛋白修饰ufi序列;(5)使用常规方式纯化/提取核酸样品中的核酸组分(导致核小体和任何其他非核酸五物质的组蛋白组分的损失);(6)产生以cdna双链体形式的cfrna条形码化的dsdna模板分子,其与至少一个衔接子结合,该衔接子具有掺入其中的cfrnaufi序列,如第6部分所述;(7)产生对应于含有5hmc残基的cfdna和分别含有5mc残基且不含5hmc的cfdna的dsdna模板分子,如第8部分所述;(8)扩增和合并以上产生的所有dsdna模板分子,其中扩增可以在合并之前或之后进行;(9)对所得扩增子的混合物进行测序;以及(10)对序列读取进行反卷积以测定关于原始生物样品的多种类型信息,包括(a)蛋白质分析物的身份和浓度、(b)组蛋白修饰的身份和位置、(c)cfrna序列信息、(d)cfdna的羟甲基化模式;和(e)cfdna的甲基化模式。本发明的具有一个或两个较少分析的组合工作流程方法在图4和图5中示意性地示出。10.ufi序列在组合工作流程方法中的意义:将理解的是,本发明的显著优点在于使用基于经典测序的技术来测定生物样品的一个或更多个非经典序列特征,其中“非经典序列特征”是指除了样品中的核酸分子的一级碱基(即,dna的腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶,以及rna的腺嘌呤、胞嘧啶、鸟嘌呤和尿嘧啶)的身份和顺序之外的特征。在最终序列读取中获得的经典序列信息中“编码”的目标非经典序列特征可以是与核酸组成有关的信息,例如经修饰的胞嘧啶残基(例如5hmc或5mc)的分布,或者它可以与核酸组成无关,而是关于血液样品中血浆蛋白的存在和浓度、在血液样品的无细胞核小体级分中观察到的组蛋白修饰等,如以上详细讨论的。即,该分析包括将目标非经典序列特征(例如血浆蛋白的身份;血浆蛋白的浓度;组蛋白修饰法数量、位置和类型;核酸的羟甲基化图谱;或核酸的甲基化图谱)转换为经典序列数据。所得经典序列数据包括至少一种ufi,即长度为约4至约36个碱基对的特定核酸序列,其中该ufi掺入dsdna模板分子内并且涉及生物样品的特定特征,即目标非经典序列特征,如以上所解释的。因此,在另一个实施方案中,本发明提供了一种用于测定核酸模板分子的非经典序列特征的基于测序的方法,其包括:将独特特征标识符序列附加到核酸模板分子,该独特特征标识符序列指示模板分子的特定非序列特征;扩增该核酸模板分子和附加的标识符序列,以得到多个扩增子,每个扩增子包含该附加的标识符序列;以及对该扩增子进行测序,并且从获得的序列读取中测定非序列特征。通常,核酸模板分子包含含有多个不同核酸模板分子的组合物内,并且至少一种指示每个模板分子的特定非经典序列特征的标识符序列附加于此。非经典序列特征可以包括与该核酸模板分子在某一点相关的蛋白质(例如,组蛋白)的一个方面。非经典序列特征也可以是生物样品中的特定蛋白质的存在或浓度,使用在第2部分中描述的邻近延伸测定法进行其向经典序列特征的转换。其他目标非经典序列特征包括,通过举例的方式,cfdna羟甲基化图谱和cfdna甲基化图谱。11.截短的衔接子:本发明另外地关于截短的测序衔接子及其在dsdna模板分子的扩增和测序中的用途。该截短的衔接子,与某些引物构建体结合使用,是在pcr扩增期间向dsdna模板分子中添加标识符条形码中是有用的。截短的测序衔接子是呈y-构建体形式,具有2个碱基对至50个碱基对的双链片段和两个各自包含2个碱基至25个碱基的单链片段。通常,该双链片段包含5个碱基对至35个碱基对和两个各自包含5个碱基至25个碱基的单链片段,例如分别地5个碱基对至25个碱基对和约5个碱基至20个碱基。在用于向dsdna模板分子中添加标识符条形码的方法中,首先使用常规手段将该截短的测序衔接子连接至末端钝化的a尾dsdna模板分子。然后在pcr方法中使用至少一种条形码化的引物将如此提供的连接有衔接子的dsdna模板分子扩增,其中该条形码引物包含:(i)第一区域,其不与衔接子中的任何序列互补并且包含一个或更多个标识符条形码;和(ii)第二区域,其与衔接子的单链片段充分互补以与其杂交,使得延伸部在在聚合酶的存在下条形码化引物的延伸产生引物的第二区域和衔接子的单链片段的双链复合物,其中包含标识符条形码的第一区域延伸超出双链复合物的末端作为单链寡核苷酸尾部。使用截短的衔接子是在本文实验部分中举例说明。尽管衔接子与dna模板分子之比可以变化,但是该比率通常为约1:5至约250:1(w/w),例如5:1至200:1、10:1至150:1或20:1至100:1。本发明还提供一种用于扩增和测序dsdna模板分子的试剂盒,其包含:(a)呈y-构建体形式的测序衔接子,其具有包含2个碱基对至50个碱基对的双链片段和两个各自包含2个碱基至25个碱基的单链片段;(b)条形码化的引物,其包含(i)第一区域,其与所述衔接子中的任何序列不互补并且包含标识符条形码;(ii)第二区域,其与所述衔接子的单链片段充分互补以与其杂交;和(c)聚合酶。在本文的实施例中详细描述了使用截短的衔接子的方法。代表性的截短的衔接子在图7中示出,并且可以与图7所示的现有技术的标准衔接子比较。图8示意性地示出了在pcr方法中使用的索引引物和截短的衔接子。截短的衔接子方法可以与本发明所述的任何方法组合,包括在扩增和测序之前将dna衔接子连接至双链dna模板分子。12.采用其他方法的实施:重要的是,本文详细描述的本发明的方法可以与包括对生物样品测序的传统技术组合。例如,本发明的方法可以与包括基于序列的富集的传统(或以后发现或开发的)液体活检方法组合,例如当选择性捕获(即,使用杂交捕获)和/或选择性扩增(例如,使用多重pcr扩增子或多种引物rca)特定基因或“热点”。基于序列的富集步骤或基于多种序列的富集步骤,例如在靶向测序的背景下,可以与本发明的方法中的任一种结合通过以下进行:通过基于序列分离一组或更多组条形码化模板分子或其扩增产物,分析该组,以及任选地在组合测序步骤之前将该组与其他核酸级分重组或合并。即,在使用例如多重pcr和阵列(其中杂交探针掺入了过程条形码和/或其他ufi序列,允许在没有直接测序情况下区分特定基因座中的标记)的更多离散数目的靶基因座的分析中,本发明的方法及其方面可以与非鸟枪测序技术组合。可以与本发明的方法或其方面组合的靶向测序方法的实例包括在以下文献中描述的那些:so等(2018)genomicmedicine3:2;gong等,上文引用;stahlberg等(2016)nucacidsres44(11;e105)1-7;mamanova等(2010)naturemethods7(2):111-118;以及其他文献。前述出版物通过引用并入本文。根据实验:当前5hmc的富集和全基因组测序(wgs)的工作流程完全并行。在实施例1-3中,评价了替代的测序衔接子构建体,其允许单个模板连接反应,其可以等分到5hmc富集或wgs并且仅在pcr扩增时通过样品索引。替代的衔接子(长度为约6个碱基至约30个碱基的单链尾)是“截短”的衔接子,其与经修饰的索引pcr序列配对。实施例1描述了定制衔接子的制备,包括衔接子序列的设计和衔接子构建体的产生;实施例2描述了用实施例1中制备的衔接子文库制备的优化;并且实施例3提供了相对于在标准可商购获得的“y”衔接子中看到的富集性能,定制衔接子的至少等效的5hmc富集性能的验证。图6(现有技术)示出了用于illuminay-衔接子构建体的标准衔接子配置;如在图中可以看到的,样品索引条形码(表示为[索引])包含在y-衔接子内。图7示出了本发明的截短的衔接子配置,在扩增引物内具有样品索引条形码(表示为[索引rc])。在两个图中突出显示了扩增引物。根据本发明使用截短的衔接子和索引引物的索引文库的创建在图8中示意性地示出。用于5hmc富集、文库生成和高通量测序的方案:在受试者入组时从通过常规静脉放血术获得的全血标本中分离出血浆。根据制造商的协议(streck,lavista,ne)(https://www.streck.com/collection/cell-free-dna-bct/),在cell-free管中收集全血。将试管维持在15℃至25℃,其中在放血的24小时内通过将全血在rt下以1600xg离心10min进行血浆分离,然后将血浆层转移到新的试管中用以16,000xg离心10min。将血浆等分用于随后的cfdna分离或在-80℃下存储。在收集核酸之前,将4ml血浆体积裂解30分钟;将所有cfdna洗脱液收集在60μl体积缓冲液中。按照制造商的协议,使用qiaamp循环核酸试剂盒(qiagen,germantownmd)分离cfdna。使用dnamini试剂盒(qiagen)提取全血基因组dna,并使用dsdna片段化酶(neb)进行片段化。通过bioanalyzerdsdna高灵敏度测定法(agilenttechnologiesinc,santaclara,ca)和qubitdsdna高灵敏度测定法(thermofisherscientific,waltham,ma)对dna进行定量。加标扩增子制备:为了产生加标对照,通过taqdna聚合酶(neb)对λdna进行pcr扩增,并通过ampurexp珠(beckmancoulter)在不重叠的约180bp扩增子以及datp/dgtp/dttp和以下中的一种的混合物中进行纯化:dctp、dmctp或10%dhmctp(zymo)/90%dctp。引物序列如下:dctpfw-5′-cgtttccgttcttcttcgtc-3′、rv-5′-tactcgcaccgaaaatgtca-3′;dmctpfw-5′-gtggcgggttatgatgaact-3′、rv-5′-cataaaatgcggggattcac-3′;10%dhmctp/90%dctpfw-5′-tgaaaacgaaaggggatacg-3′、rv-5′-gtccagctgggagtcgatac-3′。5-羟甲基胞嘧啶测定富集:如前面所述的(song等(2017)cellresearch27:1231-1242)进行测序文库制备和5hmc富集,其通过引用并入本文。对于每个测定,将cfdna标准化为10ng总输入,并且连接至测序衔接子。通过两步化学法将5hmc碱基生物素化,随后通过与dynabeadsm270链霉亲和素((thermofisherscientific,waltham,ma)结合来富集。通过bioanalyzerdsdna高灵敏度测定法(agilenttechnologiesinc,santaclara,ca)和qubitdsdna高灵敏度测定法(thermofisherscientific,waltham,ma)对所有文库进行定量,并且在测序制备中进行标准化。dna测序和比对:根据制造商的建议,使用具有第2版试剂化学法的nextseq550仪器用75个碱基对的末端配对测序进行dna测序。每个流通池对24个文库进行测序,并且使用illuminabasespacesequencehub进行原始数据处理和多路分解,以生成样品特定的fastq输出。使用具有默认参数的bwa-mem将测序读取与hg19参考基因组进行比对(li&durbin(2010),"fastandaccuratelong-readalignmentwithburrows-wheelertransform,"bioinformatics26:589-595)。峰检测:采用bwa-mem读取比对来鉴定密集读取积累的区域或峰,这些区域或峰标记了cpg内容物中羟甲基化胞嘧啶残基的位置。在鉴定峰之前,对包含比对读取位置的bam文件进行过滤用以查找不良映射(mapq<30)和未正确配对的对数。使用macs2(https://github.com/taoliu/macs)进行5hmc峰调用,p值截止=1.00e-5。如其他地方定义的,经鉴定的5hmc峰位于“黑名单区域”中。(https://sites.google.com/site/anshulkundaje/projects/blacklists)和x、y染色体和线粒体基因组的读取日期也被删除。使用具有默认参数的homer软件(http://homer.ucsd.edu/homer/)进行基因组特征富集重叠5hmc峰的计算。实施例1衔接子序列的设计和衔接子构建体的产生:表1中的定制寡核苷酸(从idt,integrateddnatechnologies,coralville,ia获得)包括三个子集:(1)用于杂交和产生衔接子构建体的截短的衔接子寡核苷酸;(2)用于扩增连接有衔接子的产物和掺入样品索引的索引pcr寡核苷酸;(3)用于再扩增含有任何索引基序的文库的通用pcr寡核苷酸。对于初始测试,创建了24个独特索引。索引来自一组可商购获得的索引,并且被检测为索引pcr寡核苷酸引物中序列的反向互补序列(索引1_引物=caagcagaagacggcatacgagatgtcggtaagtgactggagttcagacgtgtgctcttccgatc*t索引x_引物=caagcagaagacggcatacgagataggtcactgtgactggagttcagacgtgtgctcttccgatc*t;使用相同方法制备另外的索引引物)。表1:重构的200μmp5截短的和p7截短的衔接子寡核苷酸如下杂交。在1.5ml的微量离心管中产生在ste缓冲液中截短的衔接子寡核苷酸的主混合物,并等分到三个0.2ml薄壁pcr管中(每个40μl)。在以下条件下,在带有加热盖(105℃,块温度>40℃,否则为环境温度)的eppendorfmastercyclerpro上杂交寡核苷酸:将杂交的衔接子合并到单个1.5ml的微量离心管中,并通过验证在4℃下存储(建议在-20℃下长期存储)。将衔接子在1xste缓冲液中以1:250稀释,并在bioanalyzer高灵敏度芯片上进行评价。在衔接子的电泳描记图中可见一个大峰,表明杂交成功。以上程序通常适用于索引序列的产生,并且可以以一种或更多种方式被修饰,这对于本领域普通技术人员将是显而易见的。实施例2文库制备的优化:该实施例描述了使用如实施例1中所述的制备的截短的衔接子优化文库制备。(a)用于定制衔接子评价的模板dna的制备:模板dna在实际中受到限制,因此出于优化和验证截短的衔接子的目的,片段化的基因组dna被认为为可获得大量均质的dna模板提供了最佳的解决方案。出于此目的,使用了hyperplus试剂盒(roche);尽管hyperplus试剂盒通常用于组合的片段化和文库制备(包括衔接子连接),但是该实施例仅使用片段化部分。在缓冲液tris-hcl(ph8.0)溶液中,将脑和脾基因组dna储备液稀释至500ng/35μl。每种组织制备两个重复配制品,总1μg基因组dna/每种组织类型。对于脑gdna和脾gdna二者,浓度和反应体积如下:储备液浓度,250ng/μl;终浓度,10.7ng/μl;反应体积,1.5μl;eb缓冲液,33.5μl。将片段化缓冲液和酶在冰上解冻,并在0.2ml薄壁pcr管中添加到每个基因组dna样品中。片段化反应混合物的浓度和1x反应体积如下:dsdna储备液浓度,10.7ng/μl;dsdna反应浓度,7.5ng/μl;dsdna1x反应体积,35μl;片段化缓冲液储备液浓度,10x;裂解缓冲液反应浓度,1x;片段化缓冲液1x反应体积,5μl;片段化酶储备液浓度,5x;片段化酶反应浓度,1x;片段化酶1x反应体积,10μl。然后在以下条件下,将基因组dna样品在eppendorfmastercyclerpro(加热盖关闭)上进行片段化:冷却,在4℃持续1分钟;片段,在37℃下持续35分钟。立即从热循环仪中取出片段化样品,并使用如下所述的2x比率ampurexp珠协议进行纯化:在纯化之前将ampurexp珠温热到室温至少30分钟;通过将4ml超纯乙醇与1ml分子级水混合制备了80%乙醇溶液;将每个片段化样品的全部体积转移到标记好的1.5ml试管中;向每个样品中添加100μlampurexp珠并短暂涡旋以混合;在室温下使dna与珠结合10分钟;将管置于磁力架上,并允许珠粒>1分钟,然后取出并丢弃上清液;在管仍在磁力架上的情况下,向每个样品中添加500μl80%乙醇,并在磁力架上孵育30秒,然后取出并丢弃上清液;使珠粒在室温下用不加盖的样品管干燥5分钟;向每个样品中添加52μl缓冲液eb,并上下吸移以完全溶解珠粒。使样品在室温下洗脱到溶液中5分钟,偶尔混合;将管置于磁力架上,并使珠粒>1分钟;将50μl上清液小心地转移到标记好的1.5ml管中。丢弃剩余的管和珠。使用qubitdsdna测定法对片段化的dna进行定量。评价1μl的每个样品。在bioanalyzer高灵敏度芯片上评价片段化的dna样品的大小分布;所获得的大小分布曲线在图9中示出。基因组dna样品的片段化是成功的,在脾gdna配制品中的产率比在脑gdna配制品中的产率略高。观察到的片段大小在以167bp为中心的标准cfdna大小分布范围内。片段化gdna的产率足以(1)向文库制备中输入滴定量的衔接子和(2)截短的衔接子与标准衔接子的头对头评价。(b)衔接子滴定度和连接:在跨越约5倍至约500倍的衔接子与模板dna之比(5:1;20:1;50:1;100:1;250:1和500:1)的50倍范围内,将衔接子滴定到用于脑(10ng输入)和脾(20ng输入)片段化的cfdna模板的wgs文库配制品中。将片段化的dna以50μl体积标准化为10ng(脑)或20ng(脾)。制备了末端修复和a-尾酶混合物,并在eppendorfmastercyclerpro(加热盖)上,使用以下条件对片段化的基因组dna进行末端修复和a-尾化:在20℃末端修复30分钟;在65℃下热灭活30分钟;并保持4℃。将1.5μl的9nm、36nm、91nm、182nm或455nm浓度的衔接子稀释液添加到每个经末端修复的样品中,并在添加连接主混合物之前进行混合。制备具有以下组分的连接主混合物:在室温下将经末端修复的片段化的gdna样品与衔接子连接持续30分钟,并使用标准1.2x比率的ampurexp珠协议将经连接的产物纯化。(c)扩增和验证:制备pcr主混合物,其以10:1:1:2:6的体积比包含以下组分:2xkapahifihotstartreadymix;10μm通用引物;10μm索引引物(1-10);经连接的dna;和hplc水。pcr循环条件如下:初始变性,98℃,持续45秒;变性,98℃,持续15秒;退火,60℃,持续30秒。如前所述,使用标准1.2xampurexp珠协议将经扩增的产物纯化。将经pcr扩增的文库在缓冲液eb中稀释25x,并在bioanalyzer高灵敏度芯片上和用qubit高灵敏度dsdna测定法进行评价。数据如下所示。截短的衔接子滴定度文库浓度:衔接子脑文库脾文库9nm1.1ng/ul1.3ng/ul36nm5.1ng/ul10.7ng/ul91nm6.6ng/ul15.8ng/ul182nm9.4ng/ul25.3ng/ul将wgs文库浓度(ng/μl)相对于衔接子输入浓度以及mt组wgs作图,如图10所示。数据显示出(1)如所预期的,文库浓度随衔接子浓度而大大增加,和(2)在该实验中,在连接反应中182nm衔接子浓度或六倍稀释的浓缩衔接子储备液,文库浓度是最佳的。然而,由于在每个数据点上进行单次复制,因此变化未知。以前,加工用于wgs的样品是从10ngcfdna制备的;在该实验中,制备了20ng的gdna,但是仅20%用于全基因组分析,或使用标准y-衔接子的现有方法的wgs输入dna的40%的对等物。图10所示的参考值反映了针对该40%输入调整校正的所有wgs库浓度。这些数据的含义是,与用20ng输入dna的截短的衔接子连接比现有化学方法更有效地采样模板分子。基于这些分析,六倍稀释的浓缩衔接子储备液是最佳的。实施例3衔接子性能的头对头比较:(a)文库定量:基于初始评价,进行了截短相对于标准(bioo)衔接子的头对头评估。对于该实验,如上所述,各自一式两份地制备了20ng片段化的脑gdna和20ng脾gdna;然而,剩余的80%的连接有衔接子的gdna产物通过5hmc富集协议加工。作为比较,使用标准协议(bioo衔接子)制备了10ng的用于wgs和5hmc富集的每种dna类型。所有样品在同一流通池上测序以进行比较分析;当在bioo和定制样品索引上通过8bp索引读取时,不得不选择hamming距离>2的索引。·基于来自脑和脾样品的片段化的基因组dna的大小分布,用截短的衔接子产生的文库的大小分布非常接近预期;·bioo衔接子配制品的大小分布显著大于截短的衔接子产生的大小分布,可能是由于在清除期间使用的ampurexp珠比率不同(注意,在衔接子连接和pcr之后,使用0.8xampurexp珠比率用于bioo衔接子制备;相对于现有bioo衔接子协议的无注释变化连接后使用0.8x珠比率和pcr后使用1x珠比率);·由于bioo衔接子协议中的ampurexp珠清除条件,重要的文库可能丢失。此外,丢失的片段大小可以在100-150bp范围内富集,并且可以富含信号。文库定量数据总结如下:(b)测序:基于平均片段大小,将所有文库标准化为2nm浓度。在nextseq550仪器上使用单个8bp样品索引在配对末端(2x75bp)中对文库进行测序。高水平序列数据总结如下。注意,由于在该实验中操作员错误并且未正确解复用,一个样品(s8)未正确地被样品表中的索引标签化。在序列数据中观察到的片段大小分布紧随在生物分析仪上观察到的那些,其中对于wgs文库,观察到了显著的大小变化(其中针对使用bioo协议选择了小的片段),但是在将协议修改为工作ampurexpspri珠条件后没有显著的差异。(c)分析:将16个文库的每一个的基因计数转换为rpkm值。比较了wgs和5hmc配制品的处理过程中的rpkm值。通常,配制品彼此相对相似,尽管比较时有一些噪音。值得注意的是,来自脑基因组dna的5hmc配制品彼此明显相似,特别是在重复配制品之间。为每个文库构建rpkm值的柱状图。如预期的,wgs数据的rpkm分布狭窄,模态值接近1(读取的随机分布),并且次要分布接近0.5,对应于这些男性样品中x染色体基因体的剂量为50%。wgs文库的rpkm分布在很大程度上是一致的,脑gdna的5hmc文库rpkm分布也是一致的。然而,在脾gdna的5hmc文库rkpm分布中观察到显著的变化性。值得注意的是,两个文库似乎接近wgs配制品而不是5hmc配制品,表明这些文库的富集过程中可能有背景噪音。(d)估计的模板分子采样效率(仅wgs):在该实验中观察到的wgs文库浓度表明,在测试的条件下截短的衔接子具有比模板dna采样高约三倍的效率(平均值2.99x,中位数3.19x)。注意,采样效率的估计取决于输入模板浓度、输出文库浓度和pcr扩增效率;仅前两个值是已知的,但是保守的(出于该目的)对100%完美扩增效率的估计表明,截短的衔接子采样约25%的模板,bioo衔接子采样约8.5%的模板。头对头衔接子比较的结果在图11(所采样的模板的比例相对于假定的pcr效率的图)中示出。实施例4截短的衔接子功能:可以通过比较由单个模板制备的全基因组文库的产率与用有限pcr循环的替代衔接子策略来估计截短的衔接子和索引pcr引物的相对功能。在这种情况下,假定增加的全基因组文库浓度反映了模板分子更有效的采样,这表明在全球范围内更高效的过程。与可商购获得的衔接子(kapa,bioo)相比,在上述条件下,截短的衔接子在采样模板dna分子上更高效约1.5x-2x。此处观察到的值表明>18%(截短的衔接子)相对于>10%(bioo衔接子)将模板dna转化为文库的效率有所提高。衔接子效率的并排比较在图12中示出。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1