从循环核酸中鉴定全基因组序列数据中的全局序列特征的制作方法
本公开总体上涉及癌症筛查,且更特别地涉及用于鉴定从无细胞dna(cfdna)样品获得的全基因组序列数据中的全局癌症特异性序列特征的技术。
背景技术:
血浆基因型分型测定法和其他液体活检测定法的发展已经扩大了无细胞dna(cfdna)作为用于癌症患者管理的非侵入性癌症生物标志物的临床实用性。例如,血浆基因型分型测定法可以在由非恶性细胞脱落的野生型dna的高背景下,对循环肿瘤dna(ctdna)内临床相关的点突变、插入/缺失、扩增、重排和非整倍性进行无创地检测和定量。与传统的物理和生化方法相比,基于血液的ctdna检测提供了一种非侵入性且易于使用的方式用于监测疾病状态、判断预后和指导治疗。然而,由于血浆基因型分型测定法和其他液体活检测定法已证明在无创ctdna突变检测和微小残留病(mrd)监测方面的实用性,因此人们有兴趣扩展这项技术,以在做出临床诊断(即癌症筛查)之前确定其是否有能力来判别癌症的存在。
当前,针对cfdna的下一代测序(ngs)测定法旨在从覆盖已知癌基因和反复发生癌症突变位置的小靶标组(通常大小<300kb)中提取信息,并且此类组在监测疾病状态方面取得了成功。在一些方法中,ctdna突变已与其他多种基于血液的分析物(诸如外泌体、循环肿瘤细胞、蛋白质和代谢产物)整合在一起,以及随着时间的推移针对每个个体整合了这些信号,以扩展用于cfdna的ngs测定法来检测早期癌症。然而,对于筛查应用(在症状之前或在临床诊断之前的癌症检测),与监测相比,比起发现癌症样品与正常样品之间的有差异的cfdna序列数据中一般的全局癌症特异性序列特征,特定癌症突变的存在或不存在并不重要。因此,人们期望有用于癌症筛查的新技术。
技术实现要素:
提供了用于鉴定从无细胞dna(cfdna)样品获得的全基因组序列数据中的全局癌症特异性序列特征的技术(例如,方法、系统、存储由一个或多个处理器执行的代码或指令的非暂时性计算机可读介质)。
一台或多台计算机的系统可以配置为通过在系统上安装软件、固件、硬件或它们的组合来执行特定的操作或动作,所述软件、固件、硬件或它们的组合在操作中使系统执行这些动作。一个或多个计算机程序可以被配置为通过包括指令来执行特定的操作或动作,所述指令在由数据处理设备执行时使该设备执行这些动作。一个总的方面涉及一种方法,该方法包括:(a)通过数据处理系统从来自受试者的无细胞dna样品中获得全基因组序列数据,其中所述全基因组序列数据包括多个全基因组序列读段;该方法还包括(b)通过数据处理系统从多个基因组序列读段的至少大部分中计算两个或更多个度量,其中两个或更多个度量中的第一度量是(i)无细胞dna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡。该方法还包括:(c)通过数据处理系统将两个或更多个度量输入到分类器中以获得对第一类的第一预测和对第二类的第二预测,其中第一类为包括循环肿瘤dna的无细胞dna样品并且第二类为不包括循环肿瘤dna的无细胞dna样品。该方法还包括(d)通过数据处理系统基于第一预测和第二预测将无细胞dna样品分类为第一类或第二类。该方面的其他实施例包括记录在一个或多个计算机存储装置上的相应的计算机系统、设备和计算机程序,其各自配置为执行所述方法的动作。
实施方案可以包括以下特征中的一个或多个。该方法中,两个或更多个度量中的第二度量是:(i)无细胞dna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡;并且其中第二度量与第一度量不同。该方法中,分类器是线性判别分析。该方法中,计算第三度量并将其输入到分类器中,所述第三度量是:(i)无细胞dna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡;并且其中第一度量、第二度量和第三度量中的每一者都是不同的度量。
实施方案可以包括以下特征中的一个或多个。该方法中,通过将在样品中获得的无细胞dna片段大小进行标准化,从而获得概率密度函数值,来计算无细胞dna的片段大小。该方法中,所述无细胞dna的片段大小包括概率密度函数内的区域的比率。该方法中,所述概率密度函数内的区域的比率包括:长度为介于约116与约156个核苷酸之间的无细胞dna片段大小的概率的比率;以及长度为介于约164与约168个核苷酸之间的模式附近的无细胞dna片段大小的概率的比率。
实施方案可以包括以下特征中的一个或多个。该方法中,无细胞dna的片段大小是通过以下方式获得的统计学得分:(i)将样品中获得的无细胞dna片段大小进行标准化,从而获得概率密度函数值;(ii)确定无细胞dna片段大小的值的对数和连续无细胞dna片段大小之间的一阶差分;(iii)除去至少20个最低的无细胞dna片段大小,以获得剩余的无细胞dna片段大小;以及(iv)与包括循环肿瘤dna的无细胞dna和不包括循环肿瘤dna的无细胞dna相比,确定剩余的无细胞dna片段大小的第一主成分轴。该方法中,多个全基因组序列读段的相对读段深度通过以下方式计算:(i)对无细胞dna片段大小序列读段计数进行预处理,以获得一组标准化的无细胞dna片段大小序列读段计数;(ii)确定一组标准化的无细胞dna片段大小序列读段计数的每条染色体臂的中位读段深度;以及(iii)确定每条染色体臂的中位读段深度的最大值,以获得拷贝数扩增得分。
实施方案可以包括以下特征中的一个或多个。该方法中,预处理包括:(i)将来自各个样品的序列读段计数映射到具有预先确定大小的窗口中;(ii)基于一个或多个因素过滤每个窗口中的序列读段计数,以获得每个窗口的一组剩余的无细胞dna片段大小序列读段计数;(iii)对每个窗口中的鸟嘌呤-胞嘧啶含量和可映射性偏差进行校正;以及(iv)针对来自包括循环肿瘤dna的无细胞dna样品的序列数据,将每个窗口中剩余的无细胞dna片段大小序列读段计数进行标准化。该方法中,多个全基因组序列读段的相对读段深度通过以下方式计算:(i)映射独特的无细胞dna片段大小序列读段计数,以获得以百分位测量的无细胞dna片段大小读段计数分布;以及(ii)评估在第99百分位或以上的无细胞dna片段大小读段计数分布,以确定多个全基因组序列读段的相对读段深度,并获得拷贝数扩增得分。该方法中,多个全基因组序列读段的相对读段深度通过以下方式计算:(i)映射独特的无细胞dna片段大小序列读段计数,以获得以百分位测量的无细胞dna片段大小读段计数分布;以及(ii)确定每条染色体臂的序列读段计数深度的至少第90百分位除以每条染色体臂的中位序列读段计数深度的比率,以获得拷贝数扩增得分。
实施方案可以包括以下特征中的一个或多个。该方法中,使用统计学模型来计算种系等位基因失衡,以确定无细胞dna样品中一个或多个种系等位基因失衡位点的中位概率值,并获得等位基因失衡得分。该方法中,统计学模型包括二项式概率模型。该方法中,如果一个或多个种系等位基因失衡位点的中位概率值低于预先确定的显著性水平,则该中位概率值指示无细胞dna样品中一个或多个种系位点处的等位基因失衡。
实施方案可以包括以下特征中的一个或多个。该方法中,种系等位基因失衡包括杂合性的丧失。该方法中,在对受试者中的癌症进行临床诊断之前,从受试者获得无细胞dna样品。该方法中,在对受试者中的癌症进行临床诊断之后,从受试者获得无细胞dna样品。该方法进一步包括通过数据处理系统基于无细胞dna样品的分类为第一类还是第二类来预测受试者是否具有微小残留病。该方法进一步包括在预测该受试者确实具有微小残留病时,修改该受试者的治疗。
一个总的方面涉及一种方法,该方法包括:(a)计算从受试者的无细胞dna样品获得的全基因组序列数据的特征的两个或更多个得分,其中所述特征包括:(i)无细胞dna的片段大小,(ii)多个全基因组序列读段的相对读段深度,(iii)种系等位基因失衡,(iv)软剪切率,(v)取代类型的比率,(vi)总体预测的体细胞突变计数,(vii)不一致读段的比率,(vi)相对line/sine元件读段深度,或它们的组合;(b)通过数据处理系统将两个或更多个得分输入到分类器中以获得对第一类的第一预测和对第二类的第二预测,其中第一类是包括循环肿瘤dna的无细胞dna样品并且第二类是不包括循环肿瘤dna的无细胞dna样品;(c)通过数据处理系统基于第一预测和第二预测将无细胞dna样品分类为第一类或第二类;以及(d)通过数据处理系统基于无细胞dna样品的分类为第一类还是第二类来确定受试者是否具有微小残留病。
实施方案可以包括以下特征中的一个或多个。该方法中,将无细胞dna样品分类为第一类时,确定受试者具有微小残留病。该方法中,将无细胞dna样品分类为第二类时,确定受试者不具有微小残留病。该方法进一步包括通过数据处理系统基于受试者是否具有微小残留病来预测该受试者的治疗方案的临床结果;并且在确定受试者确实具有微小残留病并预测阴性临床结果时,修改受试者的治疗方案。该方面的其他实施例包括记录在一个或多个计算机存储装置上的相应的计算机系统、设备和计算机程序,其各自配置为执行所述方法的动作。
上文和下文所述的技术可以以多种方式和以多种背景来实施。参考以下附图,提供了几个示例实施方案和背景,如下文更详细描述的。然而,以下实施方案和背景只是其中的一些。
附图说明
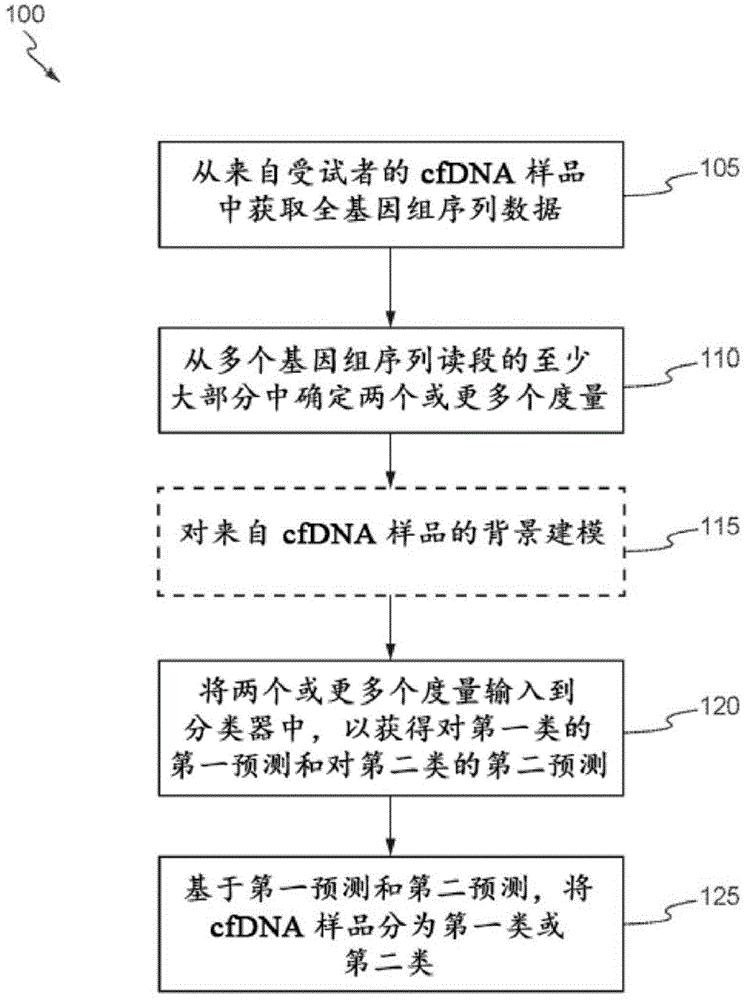
图1描绘了流程图,该流程图示出了根据各个实施例的在从无细胞dna样品获得的全基因组序列数据中鉴定全局癌症特异性序列特征的过程。
图2描绘了根据各个实施例的序列分析系统的框图。
图3描绘了根据各个实施例的计算系统或数据处理系统的框图。
图4描绘了流程图,该流程图示出了根据各个实施例的用于计算片段得分的过程。
图5描绘了流程图,该流程图示出了根据各个实施例的用于计算拷贝数扩增得分的过程。
图6描绘了根据各个实施例的预处理以从癌症样品和正常样品的覆盖率谱中去除噪声的效果。
图7a至图7c描绘了根据各个实施例的特征摘要的单变量分析,以从正常样品中分离出结肠癌和肺癌数据集。
图8描绘了根据各个实施例的用于特征摘要的多变量分析中的分类器的接收者工作特征(receiveroperatorcharacteristics,roc)曲线下面积(auc)。
图9描绘了根据各个实施例的来自健康、患有结肠癌或患有肺癌的个体的多个样品的lda得分和cna得分。
图10描绘了流程图,该流程图示出了根据各个实施例的用于诊断具有微小残留病的患者的过程。
具体实施方式
在下面的描述中,将描述各个实施例。为了说明的目的,阐述了具体的配置和细节以便提供对实施例的透彻理解。然而,对于本领域的技术人员来说也将显而易见的是,可以在没有具体细节的情况下实践实施例。此外,可以省略或简化公知的特征,以免混淆所描述的实施例。
i.介绍
在各个实施例中,提供了使用从循环核酸获得的序列数据来筛查癌症的技术(例如,方法、系统、存储可由一个或多个处理器执行的代码或指令的非暂时性计算机可读介质)。在一些实施例中,循环核酸是ctdna,其直接源自肿瘤或源自循环肿瘤细胞(ctc),其是从原发性肿瘤脱落并进入血流或淋巴系统的存活的、完整的肿瘤细胞。ctdna与cfdna不同,后者是一个更广泛的术语,它描述了在血流中自由循环但不一定是肿瘤起源的dna。因为ctdna可能反映整个肿瘤基因组,所以其潜在的临床实用性获得了关注。例如,可以以无创形式获得ctdna的液体活检,诸如在各个时间点抽血,以监测整个治疗方案中的肿瘤进展。
最近,研究人员扩展了对ctdna液体活检的常规用途,以筛查常见的癌症类型。如本文所用,针对诸如癌症的疾病或病症的“筛查”(screen或screening)是指用于确定在没有呈现症状或先前未被诊断出患有疾病或病症的受试者中该疾病或病症的可能存在或不存在的技术。这些测定可同时评估基于血液的分析物(诸如外泌体、循环肿瘤细胞、蛋白质和代谢产物)的水平以及血液中cfdna的癌症基因突变的存在。故意将用于鉴定癌症基因突变的测定中的突变组保持较小,以使假阳性结果最小化并使测定负担得起。与这些测定法相关的一个问题是,与局部疾病相比,晚期和转移性患者中循环中的ctdna更具代表性,并且随着疾病的侵袭性,发现突变的可能性增加,并因此这些测定法中的突变组并不总是灵敏的足以检测cfdna中与癌症相关的遗传变异。此外,多分析物方法对于开发具有足够灵敏度的筛查测定法至关重要,因为仅基于血液的分析物或癌症基因突变的存在不足以对筛查癌症灵敏。
为了解决这些问题,本文公开的各个实施例涉及用于鉴定从循环核酸获得的全基因组序列数据中的全局癌症特异性序列特征的技术。该技术在多变量分类器中组合了全局癌症特异性序列特征,以预测包含循环核酸的样品是否包括ctdna,并任选地保留一组参考正常值,以系统地对测序背景可变性进行建模。全局癌症特异性序列特征独立于循环核酸中存在的特定突变,并且如本文所证明,已被确定来准确地区分癌症样品和非癌症样品。令人惊讶地发现,通过这些技术鉴定出的癌症样品中的一些没有常规ctdna突变组检测到的单个体细胞突变,因此,这些技术实际上在不存在可检测到的突变的情况下使ctdna可见。
本公开的一个示例性实施例包括:(a)通过数据处理系统从来自受试者的无细胞dna样品中获得全基因组序列数据,其中所述全基因组序列数据包括多个全基因组序列读段;该方法还包括(b)通过数据处理系统从多个基因组序列读段的至少大部分中确定两个或更多个度量,其中两个或更多个度量中的第一度量是(i)无细胞dna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡。该方法还包括:(c)通过数据处理系统将两个或更多个度量输入到分类器中以获得对第一类的第一预测和对第二类的第二预测,其中第一类为包括循环肿瘤dna的无细胞dna样品并且第二类为不包括循环肿瘤dna的无细胞dna样品。该方法还包括(d)通过数据处理系统基于第一预测和第二预测将无细胞dna样品分类为第一类或第二类。如本文所用,当动作被某物“触发”或“基于”某物时,这意味着该动作被触发或至少部分地基于某物的至少一部分。
有利地,这些方法扩展了对ctdna液体活检的常规使用,以在不依赖突变检测的情况下筛查癌症。此外,已证明这些方法不仅可以准确地区分癌症样品与非癌症样品,而且还可以在未检测到单个体细胞突变的样品中检测癌症。因此,这些方法能够在不存在可检测突变的情况下使ctdna可见。
ii.从循环核酸获得的全基因组序列数据中鉴定全局癌症特异性序列特征的技术
图1阐明了用于鉴定从循环核酸获得的全基因组序列数据中的全局癌症特异性序列特征的过程和操作。各个实施例可以被描述为过程,该过程被描绘为流程图、流程简图、数据流程图、结构图或框图。尽管流程图可将操作描述为顺序过程,但许多操作也可以并行或同时执行。另外,操作的顺序可以重新布置。当一个过程的操作完成时,该过程将终止,但可能会包含未包括在附图或其描述中的额外步骤。过程可以对应于方法、函数、步骤、子例程、子程序等。当过程对应于函数时,其终止可以对应于该函数返回到调用函数或主函数。
图1中所描绘的过程和/或操作可以在由一个或多个处理单元(例如,处理器核)、硬件或它们的组合执行的软件(例如,代码、指令、程序)中来实施。该软件可以存储在存储器中(例如,在存储装置上、在非暂时性计算机可读存储介质上)。图1中特定系列的处理步骤不意图受到限制。根据替代性实施例,也可以执行其他步骤顺序。例如,在替代性实施例中,本文概述的步骤可以以不同顺序来执行。此外,在图1中阐明的各个步骤可以包括多个子步骤,这些子步骤可以按照适合于各个步骤的各种顺序来执行。此外,取决于特定的应用,可以添加或去除操作或步骤。本领域普通技术人员将认识到许多变化、修改和替代。
图1示出了流程图100,该流程图100阐明了用于鉴定从循环核酸获得的全基因组序列数据中的全局癌症特异性序列特征的过程。在一些实施例中,流程图100中描绘的过程可以由图2和图3中描绘的架构、系统和技术来实施。在步骤105,从来自受试者(例如患者)的cfdna样品获得全基因组序列数据。全基因组序列数据包括多个全基因组序列读段。可以通过单端或对端测序获得序列读段,并使用任何合适的测序技术进行分析,如第iii节中详细所述。在一些实施例中,获得一个或多个具有cfdna的样品(例如,通过从受试者抽血),通过序列分析系统进行测序,以生成用于cfdna的序列数据,并且通过数据处理系统来分析序列数据,以提供一些输出,诸如肿瘤负荷和肿瘤负荷的统计学显著性。在其他实施例中,通过数据处理系统以合适的方式从任何来源(公共或私有)获得序列数据,并通过数据处理系统进行分析以提供一些输出,诸如cfdna的片段大小、多个全基因组序列读段的相对读段深度、或种系等位基因失衡。在一些实施例中,在对受试者中的癌症进行临床诊断之前,从受试者获得cfdna样品。在其他实施例中,在对受试者中的癌症进行临床诊断之后,从受试者获得cfdna样品。
全基因组测序(也称为wgs、完全基因组测序、完整基因组测序或整个基因组测序)是一次性地确定生物体基因组的完整dna序列的过程。例如,可以通过简单的静脉穿刺从受试者中获取cfdna并用于受试者的全基因组测序。在一些实施例中,全基因组测序是低通全基因组测序以生成cfdna的低覆盖度的全基因组序列数据。如本文所用,dna测序中的“覆盖度”(或深度)是在重组序列中包括给定核苷酸的独特读段的数目。深度测序是指针对序列的每个区域的大量独特读段(例如>100x)的一般概念,并通常用于cfdna中的突变体检测。相反,如本文所用,“低通”测序是指被测序至10x以下深度的基因组。
在步骤110,从多个基因组序列读段的至少大多数中计算出两个或更多个度量。如本文所用,“大多数”是总数的较大部分或一半以上。例如,大多数是由一半以上的多个基因组序列读段组成的多个基因组序列读段的子集。在一些实施例中,两个或更多个度量中的第一度量是:(i)cfdna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡。在一些实施例中,两个或更多个度量中的第二度量是:(i)cfdna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡,并且第二度量与第一度量不同。在一些实施例中,两个或更多个度量中的第三度量是:(i)cfdna的片段大小,(ii)多个全基因组序列读段的相对读段深度,或(iii)种系等位基因失衡,并且第一度量、第二度量和第三度量中的每一者都是不同的度量。
在各个实施例中,通过对样品中获得的cfdna片段大小进行标准化来计算cfdna的片段大小,从而获得概率密度函数值。如本文所用,“片段大小”是指构成教导片段的插入物和衔接子的碱基对数目的计数或平均值。如本文所用,“插入物”是衔接子之间的碱基对,并且“插入物大小”是插入物的碱基对数目的计数或平均值。获得概率密度函数值的技术利用了ctdna和cfdna之间的表观大小差异。具体而言,以前的研究已表明ctdna高度片段化,且最常见的大小是<100bp,而正常的无细胞dna的比例更大,大小是>400bp。因此,为了检测与癌症相关的ctdna,可以使用概率密度函数值来利用片段大小的差异以将ctdna从背景cfdna分离,如第iv节中详细所述。这样,可以鉴定ctdna和cfdna之间片段长度的精确差异。在一些实施例中,cfdna的片段大小包括概率密度函数内的区域的比率。在一些实施例中,概率密度函数内的区域的比率包括:长度为介于约116个与约156个核苷酸之间的cfdna片段大小的概率的比率;以及长度为介于约164个与约168个核苷酸之间的模式附近的cfdna片段大小的概率的比率。在某些实施例中,cfdna的片段大小是通过以下方式获得的统计学片段得分:(i)将样品中获得的cfdna片段大小进行标准化,从而获得概率密度函数值;(ii)确定cfdna片段大小的值的对数和连续cfdna片段大小之间的一阶差分;(iii)除去至少20个最低的cfdna片段大小,以获得剩余的cfdna片段大小;以及(iv)与包括ctdna的cfdna和不包括ctdna的cfdna相比,确定剩余cfdna片段大小的第一主成分轴。
在各个实施例中,多个全基因组序列读段的相对读段深度通过以下方式计算:(i)对无细胞dna片段大小序列读段计数进行预处理,以获得一组标准化的无细胞dna片段大小序列读段计数;(ii)确定一组标准化的cfdna片段大小序列读段计数的每条染色体臂的中位读段深度;以及(iii)确定每条染色体臂的中位读段深度的最大值,以获得拷贝数扩增得分,如第v节中详细所述。在一些实施例中,cfdna的预处理包括:(i)将来自各个样品的序列读段计数映射到具有预先确定大小的窗口中;(ii)基于一个或多个因素过滤每个窗口中的序列读段计数,以获得每个窗口的一组剩余的cfdna片段大小序列读段计数;(iii)对每个窗口中的鸟嘌呤-胞嘧啶含量和可映射性偏差进行校正;以及(iv)针对来自包括ctdna的cfdna样品的序列数据,将每个窗口中剩余的cfdna片段大小序列读段计数进行标准化。在其他实施例中,多个全基因组序列读段的相对读段深度通过以下方式计算:(i)映射独特的cfdna片段大小序列读段计数,以获得以百分位测量的cfdna片段大小读段计数分布;以及(ii)评估在第99百分位或以上的cfdna片段大小读段计数分布,以确定多个全基因组序列读段的相对读段深度,并获得拷贝数扩增得分。在其他实施例中,多个全基因组序列读段的相对读段深度通过以下方式计算:(i)映射独特的cfdna片段大小序列读段计数,以获得以百分位测量的cfdna片段大小读段计数分布;以及(ii)评估在第99百分位或以上的cfdna片段大小读段计数分布,以确定多个全基因组序列读段的相对读段深度,并获得拷贝数扩增得分。
在各个实施例中,使用统计学模型来计算种系等位基因失衡,以获得cfdna样品中一个或多个种系等位基因失衡位点的中位概率值,并获得等位基因失衡得分,如第vi节中详细所述。在一些实施例中,统计学模型包括二项式概率模型。在一些实施例中,如果对一个或多个种系等位基因失衡位点的中位概率值低于预先确定的显著性水平,则该中位概率值指示cfdna样品中一个或多个种系位点处的等位基因失衡。在某些实施例中,种系等位基因失衡包括杂合性的丧失。
在各个实施例中,两个或更多个度量包括:(i)cfdna的片段大小,(ii)多个全基因组序列读段的相对读段深度,(iii)种系等位基因失衡,(iv)软剪切率,(v)取代类型的比率,(vi)总体预测的体细胞突变计数,(vii)不一致读段的比率,(vi)相对line/sine元件读段深度,或它们的组合。片段大小、相对读段深度和等位基因失衡可分别计算为片段得分、拷贝数扩增得分和等位基因失衡得分(如第iv、v和vi节中所述)。作为比率的度量可以被计算为属于给定类别的读段/变体(占总数)读段的百分比。在一些实施例中,两个或更多个度量中的第一度量是片段得分,并且两个或更多个度量中的第二度量是拷贝数扩增得分或等位基因失衡得分。在一些实施例中,两个或更多个度量中的第一度量是片段得分,两个或更多个度量中的第二度量是拷贝数扩增得分,并且两个或更多个度量中的第三度量是等位基因失衡得分。在一些实施例中,两个或更多个度量的确定是全基因组测序数据分析流水线的一部分,该全基因组测序数据分析流水线执行标准质量控制步骤(例如,fastq质量检查、适配器修整、重复去除)并从多个基因组序列读段中的至少大部分中计算两个或更多个度量用于下游分析。
在任选步骤115,对cfdna样品中的背景进行建模。在一些实施例中,建模包括使用在搁置的正常参考集的先验信息来鉴定“干净”的基因组区域。也就是说,例如,如果正在定义全局片段大小得分,则将首先检查一组参考法线,并且将鉴定出参考法线的区域,这些参考法线的区域的片段大小得分始终大于预先确定的阈值(诸如>200bp或>400bp)。然后可以在随后的过程中使用参考法线的区域来过滤出背景信号。可使用相同类型的预选择用于鉴定在正常情况下不存在或几乎不存在不一致性读段的区域。这样,背景信号在正常情况下保持尽可能低的水平,并提高了对cfdna样品进行分类的灵敏度和特异性。在步骤120,如第vii节中详细所述,将两个或更多个度量输入到分类器中以获得对第一类的第一预测和对第二类的第二预测。在一些实施例中,第一类是包括循环肿瘤dna的cfdna的样品,而第二类是不包括循环肿瘤dna的cfdna的样品。在某些实施例中,分类器是线性判别分析。在一些实施例中,基于来自步骤115的建模,从分类器中过滤背景。在步骤125,基于第一预测和第二预测将cfdna的样品分类为第一类或第二类。
iii.测序样品和分析系统
图2示出了根据各个实施例使用的示例性序列分析系统200,其包括在样品保持器210(例如,含有cfdna的液滴的流通池或管)内的样品205,诸如包含cfdna的血液样品。由检测器220检测来自样品205的物理特性215,诸如荧光强度值。可将来自检测器220的数据信号225发送到数据处理系统230(在检测器上或与检测器分开),该系统可包括处理器250和存储器235。数据信号225可本地存储在数据处理系统230中的存储器235中,或外部存储在外部存储器240或存储装置245中。检测器220可检测各种物理信号,诸如光(例如,来自用于不同碱基的不同探针的荧光)或电信号(例如,由穿过纳米孔的分子产生的信号)。数据处理系统230可以是或可包括计算机系统、asic、微处理器等,如关于图3进一步详细所述。数据处理系统230还可包括显示器(例如,监视器、led显示器等)和用户输入装置(例如,鼠标、键盘、按钮等)或与之耦合。数据处理系统230和其他组件可以是独立的或网络连接的计算机系统的一部分,或者它们可以直接连接到热循环仪装置或集成在其中。数据处理系统230还可包括在处理器250中执行的优化软件。基于序列数据,可以对一个或多个读段中的突变进行定量和分析,以确定肿瘤负荷和肿瘤负荷的统计学显著性。
本文所述的任何计算机系统或数据处理系统均可以利用任何合适数量的子系统。在图3中示出了计算机系统或数据处理系统(例如,参照图2所述的数据处理系统230)以及关联子系统的实例。计算系统300仅是合适的计算系统的一个实例,并且无意于对本实施例的使用或功能的范围提出任何限制。而且,不应将计算系统300解释为与序列分析系统200中所阐明的任何一个组件或组件的组合具有任何依赖性或要求。
如图3所示,计算系统300包括计算装置305。计算装置305可驻留在诸如云环境之内的网络基础设施上,或者可以是单独的独立计算装置(例如,服务提供商的计算装置)。计算装置305可包括总线310、处理器315、存储装置320、系统存储器(硬件装置)325、一个或多个输入装置330、一个或多个输出装置335以及通信接口340。
总线310允许计算装置305的组件之间的通信。例如,总线310可以是几种类型的总线结构中的任何一种,包括存储器总线或存储器控制器、外围总线和本地总线,该总线310使用各种总线架构中的任何一种来提供一个或多个有线或无线通信链路或路径用于将数据和/或功率传输到计算装置305的各个其他组件或从其之间传输。
处理器315可以是一个或多个常规处理器、微处理器或专门的专用处理器,其包括可操作以解释和执行计算机可读程序指令的处理电路,诸如用于控制用于实施本发明的功能、步骤和/或性能的计算装置305的各种其他组件中的一个或多个的操作和性能的程序指令。在某些实施例中,处理器315解释并执行本发明的过程、步骤、功能和/或操作,其可以通过计算机可读程序指令可操作地实施。例如,处理器315可以检索,例如,导入和/或以其他方式获得或生成序列数据、查询序列数据、确定或计算度量、对背景建模、确定概率值、提供(诸如类别、解释性诊断和临床结果)预测。在实施例中,可将由处理器315获得或生成的信息,例如序列数据、度量、背景模型、概率值、类别等,存储在存储装置320中。
存储装置320可包括可移动/不可移动、易失性/非易失性计算机可读介质,诸如但不限于非暂时性机器可读存储介质,诸如磁和/或光记录介质及它们相应的驱动器。根据本发明的不同方面,驱动器及其相关联的计算机可读介质提供了计算机可读程序指令、数据结构、程序模块和其他数据的存储用于计算装置305的操作。在实施例中,根据本发明的方面,存储装置320可以存储操作系统345、应用程序350和程序数据355。
系统存储器325可包括一个或多个存储介质,包括例如非暂时性机器可读存储介质(诸如闪存)、永久存储器(诸如只读存储器(“rom”))、半永久存储器(诸如随机访问存储器(“ram”))、任何其他合适类型的非暂时性存储组件或它们的任何组合。在一些实施例中,输入/输出系统360(bios)可以存储在rom中,该输入/输出系统360(bios)包括诸如在启动期间帮助在计算装置305的各种其他组件之间传递信息的基本例程。另外,数据和/或程序模块365,诸如操作系统345的至少一部分、程序模块、应用程序350和/或程序数据355,通过处理器315可以访问和/或当前对其进行操作,可能包含在ram中。在实施例中,程序模块365和/或应用程序350可以包括度量的索引或表格、算法或模型(诸如蒙特卡罗(montecarlo)算法以对背景进行建模)、分类器(诸如线性判别分析)以及比较工具(其提供用于执行处理器315的指令)。
一个或多个输入装置330可以包括允许操作员向计算装置305输入信息的一个或多个机制,诸如但不限于触摸板、拨盘、点击轮、滚轮、触摸屏、一个或多个按钮(例如键盘)、鼠标、游戏控制器、轨迹球、麦克风、相机、近距离传感器、光检测器、运动传感器、生物特征传感器及它们的组合。一个或多个输出装置335可包括向操作者输出信息的一个或多个机制,诸如但不限于音频扬声器、耳机、音频线路输出、视觉显示器、天线、红外端口、触觉反馈、打印机、或它们的组合。
通信接口340可包括使计算装置305能够与诸如移动装置或其他计算装置(诸如例如网络环境(例如云环境)中的服务器)的远程装置或系统通信的任何类似收发器的机制(例如,网络接口、网络适配器、调制解调器或它们的组合)。例如,计算装置305可使用通信接口340经由一个或多个局域网(lan)和/或一个或多个广域网(wan)连接到远程装置或系统。
如本文所讨论,计算系统300可以配置成使用血液中要监测的变体的先验知识来超灵敏地检测循环核酸。特别地,对处理器315执行包含在非暂时性机器可读存储介质(诸如系统存储器325)中的程序指令有响应,计算装置305可执行任务(例如,过程、步骤、方法和/或功能)。程序指令可从诸如数据存储装置320的另一计算机可读介质(例如,非暂时性机器可读存储介质)或经由通信接口340或云环境内部或外部的服务器从另一装置读取到系统存储器325中。在实施例中,根据本发明的方面,操作员可以经由一个或多个输入装置330和/或一个或多个输出装置335与计算装置305进行交互,以促进任务的执行和/或实现此类任务的最终结果。在额外或替代性实施例中,可以使用硬连线电路代替程序指令或与程序指令结合使用,以实施与本发明的不同方面一致的任务,例如步骤、方法和/或功能。因此,可以以硬件电路和软件的任何组合来实施本文公开的步骤、方法和/或功能。
iv.片段度量
在各个实施例中,计算与序列读段的片段大小相关的片段得分。已经发现,与健康的cfdna相比,ctdna片段中的dna片段大小更小。在一些实施例中,确定片段大小分布以计算片段大小的片段得分,如图4所示。将一个或多个cfdna样品的片段大小的分布确定为列表或函数,其显示序列数据的所有可能大小(例如,碱基对数目)以及每个片段大小出现的频率。在步骤405,对样品的片段大小分布进行标准化,使得该分布是概率密度函数(例如,描述将出现给定大小的概率的函数,以及描述给定大小或任意大小小于它的累积概率的函数是分布函数)。片段大小分布的概率密度函数可以定义为分布函数的导数。在某些实施例中,通过线性变换执行标准化,使得从序列数据获得的片段大小数据被重新缩放至单位间隔。在步骤410,对片段大小值执行对数变换(取对数),并且获得连续的插入物大小长度之间的一阶差分。这提供了有关片段分布的形状的信息。在步骤415,通过除去前20、30、40或50个片段长度值(这些计数可能太低和嘈杂)来过滤片段大小。在某些实施例中,除去10至60个片段长度值以过滤片段大小。
在步骤420,跨正常样品和癌症样品的集合计算片段长度分布的第一主成分轴。该步骤可以作为多个预处理步骤的一部分执行,以强调变化并在片段大小数据集中显示出强模式。在某些实施例中,使片段大小成为第一主要成分,并且降低第二主要成分(例如,相同片段大小的数目)。主成分分析的结果提供成分得分或因素得分(对应于特定数据点的经转换的变量值),以及负荷(应乘以每个标准化的原始变量以得到片段得分的权重)。在某些实施例中,第一主成分提供权重(负荷)以在测试数据中给出插入物大小值(参见例如,步骤105中正在处理的cfdna样品)。
在其他实施例中,片段分布的替代性总结是可能的,以增强对癌症样品的检测。例如,更低的分位数(.1%、1%、5%)的分布可用于计算片段大小的片段得分。可替代地,可以使用设定的临界值或某个范围内的概率密度函数值来计算片段大小的片段得分,例如,片段长度为120bp、130bp或140bp时的概率密度函数,或者固定数量的单元小于分布的模式(例如,对于cfdna,分布模式通常约为166bp),概率密度函数值的总和小于模式的50到10个单元之间(例如,约116bp至约156bp之间)。可替代地,概率密度函数内的区域的比率可以用于计算片段大小的片段得分,例如,介于约116bp与约156bp之间的片段长度的概率与模式周围的介于约164bp与约168bp之间的片段长度的概率的比率,因为可以预料更低片段长度的相对富集。如本文所用,术语“基本上”、“大约”或“约”可以被所指定的“在[百分比]内”所取代,其中该百分比包括0.1%、1%、5%和10%。
v.相对读段深度度量
在各个实施例中,计算拷贝数扩增得分,其与序列读段中拷贝数变化的量度相关。已经发现,与健康的cfdna相比,在ctdna片段中发现拷贝数扩增(拷贝数改变或变化增加)的存在更为频繁。相对读段深度旨在评估cfdna样品内焦点或广泛拷贝数变化的存在。因此,如本文所用,“相对读段深度”是拷贝数变化的量度。在一些实施例中,计算多个全基因组序列读段的相对读段深度,如图5所示。在一些实施例中,该计算包括步骤505,其中执行多个预处理步骤以从覆盖率谱中去除噪声并获得一组标准化的cfdna片段大小序列读段计数。在步骤505a,将来自各种cfdna样品(例如,包括ctdna的cfdna和不包括ctdna的cfdna)的序列读段计数映射到具有预先确定大小的容器或窗口中。序列读段计数是在测序中使用的针对每种探针的读段的数目,并且可以任选地根据一个或多个不同的已知因素进行校正以校正任何偏差。在某些实施例中,容器或窗口大小介于10kb与10000kb之间,例如200kb。在步骤505b,基于一个或多个因素在每个窗口中对序列读段计数进行过滤,以获得每个窗口的一组剩余的cfdna片段大小序列读段计数。过滤包括从后续分析中删除序列读段计数。在一些实施例中,一个或多个因素包括小于预先确定的阈值的序列计数。在某些实施例中,预先确定的阈值小于350个序列读段,例如,小于200个序列读段。在一些实施例中,一个或多个因素包括着丝粒读段。在一些实施例中,一个或多个因素包括具有可变细胞带的序列读段。
在步骤505c,在每个窗口中对鸟嘌呤-胞嘧啶(gc)含量和可映射性偏差进行校正。gc含量偏差描述了片段计数(读段覆盖度)与测序数据中发现的gc含量之间的相关性。如果不进行校正,则专注于测量基因组内片段丰度的分析,gc偏差可以主导目标信号。读段映射过程生成区域可映射性偏差。由于映射到基因组中多个位点的序列读段通常被丢弃,因此具有高序列简并性的基因组区域显示了映射的读段覆盖度比独特区域更低,如果不进行校正则会产生系统性偏差。在步骤505d,针对来自包括ctdna的cfdna样品的序列数据,将每个窗口中剩余的cfdna片段大小序列读段计数进行标准化。所得的清除物示出于图6中的癌症样品和正常样品中。
预处理完成后,可以根据标准化的深度数据(即,一组标准化的cfdna片段大小序列读段计数)评估多个基因组范围的摘要。在一些实施例中,标准化的深度数据的总结是染色体臂对中位标准化深度的最大值。例如,相对读段深度的计算可以进一步包括步骤510,其中针对一组标准化的cfdna片段大小序列读段计数确定每条染色体臂的中位读段深度,以及步骤515,其中确定每条染色体臂的中位读段深度的最大值以获得拷贝数扩增得分,该拷贝数扩增得分记录臂水平扩增。在其他实施例中,标准化的深度数据的总结是合并或开窗值的高百分位,诸如第99百分位、第99.9百分位和第99.99百分位。多个全基因组序列读段的相对读段深度可通过以下方式计算:(i)映射独特的cfdna片段大小序列读段计数,以获得以百分位测量的cfdna片段大小读段计数分布;以及fii)评估在第99百分位或以上的无细胞dna片段大小读段计数分布,以确定多个全基因组序列读段的相对读段深度,并获得拷贝数扩增得分。在其他实施例中,标准化的深度数据的总结是高百分位除以每条染色体臂的中位深度的比率,以便鉴定聚焦扩增。例如,每条染色体臂的深度的第90百分位除以每个臂的中位深度。多个全基因组序列读段的相对读段深度通过以下方式计算:(i)映射独特的cfdna片段大小序列读段计数,以获得以百分位测量的cfdna片段大小读段计数分布;以及(ii)确定每条染色体臂的序列读段计数深度的至少第90百分位除以每条染色体臂的中位序列读段计数深度的比率,以获得拷贝数扩增得分。
vi.种系等位基因失衡度量
在各个实施例中,计算与序列读段中拷贝数变异的量度相关的等位基因失衡得分。已经发现,与健康的cfdna相比,在ctdna片段中发现种系失衡存在的频率更高。对于正常拷贝数区域中的杂合单核苷酸多态性(snp),预期等位基因频率(af)为50%。对于具有拷贝数增加或减少的区域,af可能会偏离50%,例如,如果在某个位置有3个拷贝,则杂合snp将为2/3=66%或1/3=33%af。这被称为“种系等位基因失衡”,并且基于二项式概率提供了用于计算等位基因失衡的得分的公式。在一些实施例中,使用统计学模型计算等位基因失衡得分,以获得无细胞dna样品中一个或多个种系等位基因失衡位点的中位概率值。在一些实施例中,统计学模型包括:无效假设,其中种系变体是杂合的,并且在给定的读段处看到该种系变体的概率为p0=0.5的显著性水平,然后对某个位点处的n次读段总数进行yobs非参考读段的观察,用于拒绝原假设的p值由以下等式(1-3)给出:
在该统计学模型中,为每个种系位点创建概率值(p值),并将其用作等位基因失衡得分。例如,整个样品的中位p值低表明种系失衡。可替代地,由于种系等位基因失衡应与拷贝数变化有关,因此等位基因失衡得分可定义为等位基因失衡与标准化深度的相关性,并且当低种系等位基因失衡的p值对应于高(扩增)或低(杂合缺失)标准化深度时,就会出现相关性。
vii.单变量和多变量实验与分析
针对癌症样品(25个iv期肺癌和25个转移性crc样品)的cfdna以及来自健康对照(24个样品)的cfdna的低覆盖度(平均深度范围为1至5)全基因组序列(wgs)数据执行了概念验证实验和分析,以评估潜在的全局序列读段特征,这些读段特征表明存在癌症衍生的cfdna。尽管这些是晚期癌症,但从相同血浆样品的匹配的深度测序数据集中推断出的ctdna含量表明ctdna含量范围很广(具有<0.5%ctdna样品的15%-avenioctdna分析试剂盒的检测极限),因此它们是评估该方法的足够具有挑战性的组。开发了全基因组测序数据分析管道,该管道可以实行标准的qc步骤(fastq质量检查、适配器修整、重复删除),并计算相关的全局度量用于下游分析。
图7a至7c示出了对全局测序特征摘要进行单变量分析的能力,以从正常样品中分离出结肠癌和肺癌数据集。例如,图7a示出了肺癌、结肠癌和正常数据集之间的片段得分(pca轴1)。图7b示出了基于肺癌、结肠癌和正常数据集之间的读段深度分析的拷贝数扩增得分。图7c示出了肺癌、结肠癌和正常数据集之间的等位基因失衡得分(例如,中位p值)。如图所示,每个全局测序特征摘要都可以单独地判别癌症样品(包含ctdna的样品)与正常样品(不含ctdna的样品)。还发现本文讨论的其他特征和度量显示出判别癌症样品与正常样品之间的各种能力。
在多变量分析中,至少两个特征或度量被组合到线性判别分析分类器中,并证明了能够以更高的特异性和灵敏度来判别正常样品(不含ctdna的样品)与癌症样品(含ctdna的样品)。例如,3倍交叉验证的线性判别分析分类器用来建立性能来判别正常样品与癌症样品。图8示出了该分类器的接受者工作特征(receiveroperatorcharacteristics,roc)曲线下面积(auc),并且它显示了以100%的特异性(假阳性率=0)实现>70%的灵敏度(真阳性率)。图9示出了每个样品的线性判别分析得分和拷贝数改变得分,其通过样品中已知的体细胞单核苷酸变体(snv)的af值着色。如图所示,有一些样品被正确分类为没有可检测到的snv的癌症样品,并且还有一些snvaf<0.5%(低于lod)的样品也被可靠地分类为癌症样品。
viii.诊断测定与治疗
在各个实施例中,提供了用于通过本文公开的技术基于无细胞dna样品的分类为第一类还是第二类来确定受试者是否有微小残留病的技术。一些实施例进一步涵盖基于微小残留病的确定用于预测受试者的治疗方案的临床结果或提供受试者的癌症预后的技术。例如,一旦样品被分类为第一类或第二类,则该分类可以用于确定受试者中微小残留病的存在。
图10示出了流程图1000,其阐明了用于诊断具有微小残留病的患者的过程和操作。各个实施例可以被描述为过程,如先前关于图1所描述的,该过程被描绘为流程图、流程简图、数据流程图、结构图或框图。流程图1000中描绘的过程包括在关于图1描述的流程图100中执行的一些或全部步骤,并可以通过图2和图3中描绘的架构、系统和技术来实现。在步骤1005,从来自受试者(例如患者)的cfdna样品获得全基因组序列数据。全基因组序列数据包括多个全基因组序列读段。在一些实施例中,使用诊断测定法获得全基因组序列数据。可以以多种方式来创建测定并使用各种技术,诸如pcr、测序、杂交阵列和独特的分子标识符。该测定应能够在治疗前水平检测ctdna。在一些实施例中,可以将测定法创建为试剂盒的一部分,该试剂盒包含从cfdna获得全基因组序列数据所必需的试剂。例如,试剂盒可包含对全基因组序列具有特异性的寡核苷酸,诸如探针和扩增引物。在一些实施例中,试剂盒进一步包含用于扩增和检测测定的性能所必需的试剂,诸如pcr、实时pcr或转录介导的扩增(tma)的组分。在一些实施例中,全基因组寡核苷酸被可检测地标记。在此类实施例中,试剂盒包含用于对标记物进行标记和检测的试剂。例如,如果寡核苷酸用生物素标记,则试剂盒可包含具有酶及其显色底物的链霉亲和素试剂。
在步骤1010,计算从无细胞dna样品获得的全基因组序列数据的特征的两个或更多个得分,并将其输入到分类器中以获得对第一类的第一预测和对第二类的第二预测。在一些实施例中,特征包括cfdna的片段大小、多个全基因组序列读段的相对读段深度、全基因组序列读段的种系等位基因失衡或它们的组合。在其他实施例中,特征包括(i)cfdna的片段大小,(ii)多个全基因组序列读段的相对读段深度,(iii)种系等位基因失衡,(iv)软剪切率,(v)取代类型的比率,(vi)总体预测的体细胞突变计数,(vii)不一致读段的比率,(vi)相对line/sine元件读段深度,或它们的组合;在一些实施例中,第一类是包括循环肿瘤dna的无细胞dna样品,而第二类是不包括循环肿瘤dna的无细胞dna样品。在步骤1015,基于无细胞dna样品的分类为第一类还是第二类来确定受试者是否具有微小残留病。微小残留病是在治疗期间或治疗后保留在受试者体内的残留肿瘤的存在。例如,如果无细胞dna样品被分类为第一类,其中无细胞dna样品包括循环肿瘤dna,则可以确定受试者具有微小残留病。可替代地,如果无细胞dna样品被分类为第二类,其中无细胞dna样品不包括循环肿瘤dna,则可以确定受试者不具有微小残留病。
在步骤1020,基于受试者是否具有微小残留病来预测该受试者的治疗方案的临床结果。几项研究已经证实了评估治疗方案期间或治疗方案之后可能存在的微小残留病的重要性,以帮助预测患者的临床结果。例如,没有表现出持续的微小残留病的患者,表现显著好于表现出持续的微小残留病的患者。在步骤1025,在确定受试者确实具有微小残留病并预测阴性临床结果时,可以修改受试者的治疗方案。可替代地,在确定受试者不具有微小残留病并预测阳性临床结果时,可以维持受试者的治疗方案。
- 还没有人留言评论。精彩留言会获得点赞!