艾日布林及其中间体的制备方法与流程
本发明属于医药化学技术领域,具体涉及药物合成领域,更具体的涉及一种艾日布林及其中间的体制备方法。
背景技术:
甲磺酸艾日布林由日本卫材制药公司开发,注射液于2010年11月获得美国fda批准上市,商品名halaven。作为具有全新作用机制的微管蛋白聚合抑制剂,甲磺酸艾日布林是第1个用于转移性乳腺癌患者获得总生存期改善的单药化疗药,为局部晚期乳腺癌或转移性乳腺癌患者提高生存率和生活质量提供了新的治疗手段,是一个极具应用价值的药物。其化学结构为:
从艾日布林的结构看,分子中含有19个手性中心,合成非常困难,通常是通过如下三个片段a(c27-c35)、b(c14-c26)和c(c1-c13)拼接而成。在各片段拼接时,因离去基团的不同而有不同的路线。
cn101899026b公开了艾日布林中间体化合物er-806058与苯基磷酸二乙酯反应制备化合物er-806059,然后与tmsi反应制备化合物er-806060,最后通过醋酸硼氢化钠还原得到目标化合物er806061,该方法制备的化合物er806061为油状物,不容易纯化和保存,此外,三步反应产物均不容易纯化,都是直接投入下一步反应,不容易控制投料比,整体反应收率较低,三步反应总收率低于70%,前后各加一步,总共五步的收率为57%。
中国专利cn104876896a公开了35位碳以苯磺酰基保护的片段a与片段b拼接,再与片段c拼接。
上述方法中,片段a和b拼接时,需要加入手性配体,以片段b计,拼接反应的收率59.2%,然后再与片段c拼接,制备艾日布林的总收率仍然较低,并且纯化方法繁琐。
中国专利cn1216051c公开了35位碳以mpm(甲氧基苯硫基甲基)保护的片段a与片段b拼接,再与片段c拼接。
上述方法中,片段a和b拼接后,得到c27非对映体混合物,比例约为3:1,混合物通过多次分离纯化,以片段b计,a与b拼接反应的收率仅33.6%。mpm基团离去后与片段c拼接,而mpm的基团离去需要多次转化,步骤繁琐,收率低。
鉴于此,本发明所要解决的技术问题在于提供一种操作简便,收率高,易于固化和保存的艾日布林的新的中间体及其制备方法。
技术实现要素:
为达到上述目的,本发明提供了一种制备艾日布林的新的中间体nt027u06,以及该中间体nt027u06的制备方法;本发明还提供了一种艾日布林或其盐的制备方法。
首先,本发明提供了一种由化合物nt027b04制备艾瑞布林中间体化合物nt027u07的方法,包括以下步骤:
(1)使联苯砜基磷酸二乙酯与化合物nt027b04进行wittig-honor反应,得到的化合物nt027u05:
(2)化合物nt027u05脱去苄基,转化成化合物nt027u06:
(3)化合物nt027u06的通过还原反应,转化成化合物nt027u07:
上述方法中,优选,步骤(1)反应结束后,通过用乙酸乙酯萃取,得到粗品后,直接用于投入下一步反应;
进一步的优选,步骤(2)化合物nt027u05在三氯化硼条件下脱去苄基,转化成化合物nt027u06;
进一步的优选,步骤(3)所述还原反应是在四丁基氯化铵/三乙酰氧基硼氢化钠条件下进行;
更进一步的优选,步骤(1)在氮气保护下,将化合物nt027u00加入到thf中,搅拌,降温至0~10℃,然后滴加lhmds,然后在5~15℃继续搅拌,作为试剂a;
将化合物nt027b04加入到甲苯中,温度控制在5~15℃,然后将配好的试剂a慢慢滴加到nt027b04的甲苯溶液中,反应结束后,将反应温度降低至-5~5℃,缓慢滴加盐酸调节ph<7,然后用乙酸乙酯萃取,任选的再用饱和碳酸氢钠洗涤,饱和食盐水洗涤,分离有机层,干燥得到粗品nt027u05,可以直接用于下一步反应。
步骤(2)在氮气保护下,将化合物nt027u05溶于二氯甲烷,将反应釜降温到-70℃~-60℃时,慢慢滴加三氯化硼溶液,滴加过程中,保持温度在-60℃以下,滴加完毕后,保持反应温度在-60℃~-50℃,至反应结束;进一步的优选,反应结束后,取反应液溶液加入饱和nahco3和乙酸乙酯的混合溶液淬灭,然后向反应液中加入饱和的nahco3水溶液,搅拌,产生的固体过滤得到母液,再将母液静置分层后得到有机层,将得到的有机层用饱和碳酸氢钠溶液和饱和食盐水洗涤,用无水硫酸钠干燥干,烝除有机溶剂得到黄色油状物,粗品直接用于下一步反应。
上述方法中,进一步的优选,步骤(3)在氮气保护下,向反应瓶中加入三乙酰氧基硼氢化钠,四丁基氯化铵,dme和甲苯搅拌均匀,然后加热到60~80℃,反应持续搅拌1~2小时,然后加入nt027u06和甲苯的混合液,然后升温到75~85℃反应至结束;优选的,反应结束后,将反应液降温至-5~5℃,然后加入水,静置分层,分离有机相用饱和食盐水和水洗涤,得到的有机相用无水硫酸钠干燥,过滤后有机层旋干得黄色泡状固体,为粗品nt027u07。
进一步的,所述步骤(3)进一步包括,将粗品nt027u07溶于乙酸乙酯中,冷至-20~-10℃后缓慢滴加石油醚,析出固体,过滤收集固体,得到淡黄色固体产物,其中,乙酸乙酯与石油醚的用量为1:3~4(v/v)。
第二方面,本发明还提供了一种艾日布林或其盐的制备方法,所属方法包括采用本发明前述方法制备化合物nt027u07。
第三方面,本发明还提供了如下化合物:
与现有技术相比,本发明提供了一种制备艾日布林全新的合成方法和中间体,特别是使用联苯砜基磷酸二乙酯代取之前文献报道的苯基磷酸二乙酯化合物与nt027b04进行wittig-honor反应,反应结束后,后处理得到的粗品nt027u05直接用于下一步反应;以化合物nt027u04为起始物料经过三步反应后得到nt027u07,前两步的粗品直接用于下一步反应,三步总收率为不低于85.5%。
其次,与文献相比,本发明制备的艾日布林中间体nt027u07为固体,并且可以通过重结晶进行纯化,而文献对应的化合物为油状物。
第三,本发明步骤(2)中用三氯化硼代替了文献中的tmsi,优化了反应结果,简化了反应后处理操作。
附图说明
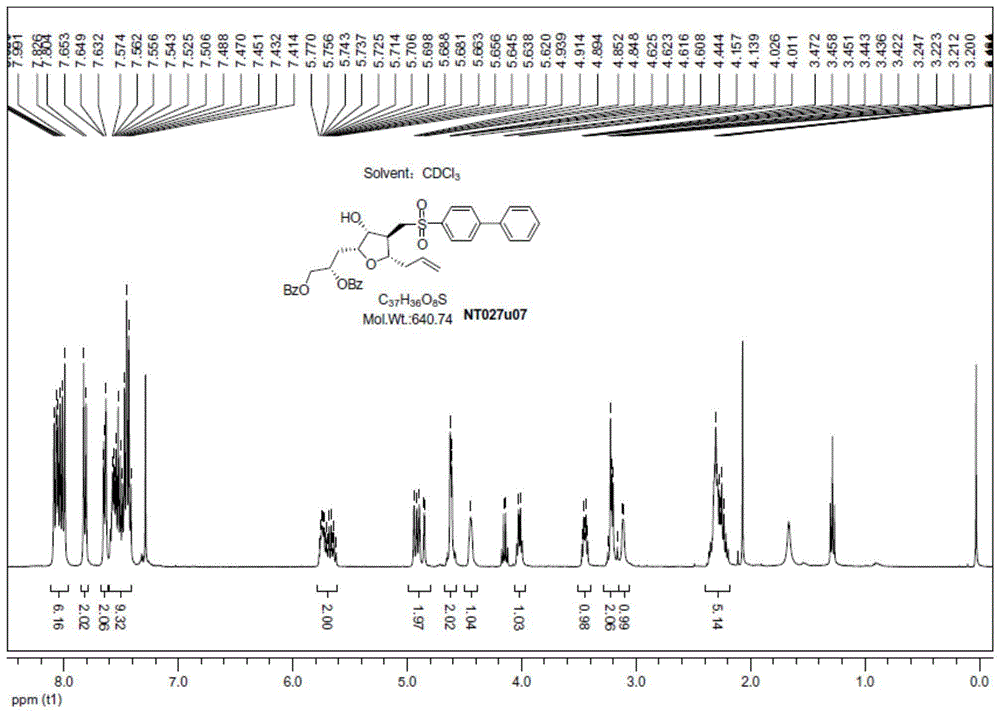
图1显示本发明方法制备的化合物nt027u07的1h-nmr谱图。
图2显示本发明方法制备的化合物nt027u07的hplc谱图,其中化合物nt027u07的纯度为98.8%。
具体实施例
以下结合具体实施例对本发明的技术方案以及优选实施方式做进一步解释和说明。
实施例1:化合物nt027u05的制备
在1l的反应瓶中,在氮气保护下,加入47g化合物nt027u00和150mlthf,搅拌溶解后,将反应体系降温,当反应温度降到5℃时,然后滴加118mllhmds(1.0m),反应液保持在10℃继续搅拌,作为试剂a。
接着将63g化合物nt027b04和300ml甲苯的溶液加入另外一个1l反应瓶中,当温度降到10℃时,然后将以上配好的试剂a慢慢滴加到此反应液中,反应1h后取样。
将反应瓶温度降到0℃,然后缓慢滴加1m的盐酸溶液至ph<7,然后萃取用ea100ml*4,再用100ml饱和碳酸氢钠洗涤一次,再用饱和食盐水100ml洗涤,分离后有机层用无水硫酸钠干燥,过滤后有机相旋干后粗品直接用于下一步反应。
实施例2化合物nt027u06的制备:
2l的反应瓶中,在氮气保护下,将实施例1获得的67.7g化合物nt027u05的粗品溶于0.3l二氯甲烷,搅拌溶解后,将反应釜降温,当反应温度降到-70℃时,慢慢滴加551ml1m的三氯化硼溶液,滴加过程中,保持温度在-60℃以下,滴加完毕后,保持反应温度在-40℃,tlc监控反应进程;
向反应液中加入饱和nahco3和乙酸乙酯(ea)的混合溶液淬灭后tlc监控反应进程,用pe:ea=4:1作为展开剂,原料rf≈0.4,产物rf≈0.3。向反应液中加入300ml饱和的nahco3水溶液,搅拌0.5小时,产生的固体过滤得到母液。静置分层后得到有机层,再用饱和碳酸氢钠溶液150ml洗涤,然后用饱和食盐水150ml洗涤一次,用无水硫酸钠干燥。过滤后有机层旋干得黄色油状物,粗品直接用于下一步反应。
实施例3化合物nt027u07的制备
在1000ml的反应瓶中,在氮气保护下,分批加入965g的三乙酰氧基硼氢化钠,91g四丁基氯化铵,300ml的dme和150ml甲苯,反应液搅拌均匀,然后加热到75℃,反应持续搅拌1小时,然后加入47g实施例2制备的nt027u06粗品和150ml甲苯的混合液,然后升温到85℃继续反应2小时,取反应液溶液tlc监控,采用pe:ea=3:1做展开剂,原料rf≈0.4,产物rf≈0.3。
将反应液降温到0℃,然后加入200ml的水,静置分层后有机相用饱和食盐水洗涤(100ml*2),再用水洗涤(100ml*2),得到的有机相用无水硫酸钠干燥,过滤后有机层旋干得黄色泡状固体。粗品溶于约20毫升乙酸乙酯中,冷至-20℃后缓慢滴加约80毫升石油醚,析出固体,继续搅拌3小时后过滤收集固体,得到40.3g淡黄色固体产物,产品纯度98.8%。以化合物nt027b04为起始物料计算,三步反应总收率85.48%。
- 还没有人留言评论。精彩留言会获得点赞!