一种高光学纯度手性氧代氮杂环烷化合物的制备方法与流程
本发明涉及一种高光学纯度手性氧代氮杂环烷化合物的制备方法,具体涉及一种高光学纯度n-p取代基-2-(r或s)-g取代基氧代氮杂环烷或其盐的制备方法,属于精细化工技术领域。
背景技术:
高光学纯度n-p取代基-2-(r或s)-g取代基氧代氮杂环烷或其盐是一种重要的中间体化合物,利用其所具备的氮原子、手性酸或其衍生物以及杂环的羰基可衍生出多种化合物,用于医药农药等的开发研究。
光学纯度n-p取代基-2-(r或s)-g取代基氧代氮杂环烷(i)或其盐,其中,n为1、2或3;具体为,当n为1时,式ⅰ化合物为光学纯度的n-p取代基-2-(r或s)-g取代基-4-氧代四氢吡咯或其盐;n为2时,式ⅰ化合物为光学纯度的n-p取代基-2-(r或s)-g取代基-5-氧代哌啶或其盐;n为3时,式ⅰ化合物为光学纯度的n-p取代基-2-(r或s)-g取代基-6-氧代杂环庚烷或其盐。式ⅰ化合物具有以下所示结构:
其中,式ⅰ化合物中,取代基p为苄基、邻甲氧基苄基、间甲氧基苄基、对甲氧基苄基、2,4-二甲氧基苄基、邻甲基苄基、间甲基苄基、对甲基苄基、邻氯苄基、对氯苄基、间氯苄基、苯甲酰基、甲氧羰基、叔丁氧羰基或苄氧羰基;取代基g为cooh或coor,其中r为甲基、乙基、正丙基、异丙基、叔丁基、正丁基、仲丁基或苄基。
式ⅰ化合物的盐具体指式ⅰ化合物的盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、磷酸盐或乙酸盐。
技术实现要素:
针对现有技术存在的不足,本发明提供一种高光学纯度手性氧代氮杂环烷化合物的制备方法,具体为一种高光学纯度n-p取代基-2-(r或s)-g取代基氧代氮杂环烷或其盐的制备方法。
术语说明:
式ii化合物:(r或s)n-p取代基-2-烷氧羰基烷基亚氨基二乙酸二酯;
式ⅰ化合物:n-p取代基-2-(r或s)-g取代基氧代氮杂环烷。
手性氧代氮杂环烷化合物:式ⅰ化合物或其盐。
本说明书中的化合物编号与结构式编号完全一致,具有相同的指代关系,以结构式为依据。
本发明的技术方案如下:
一种手性氧代氮杂环烷化合物的制备方法,包括步骤:
通过使式ii化合物经环化反应,然后经脱羧反应制备得到式ⅰ化合物或其盐;
其中,式ii化合物结构式中,取代基r1为甲基、乙基、正丙基、异丙基、叔丁基、正丁基、仲丁基或苄基;取代基p为苄基、邻甲氧基苄基、间甲氧基苄基、对甲氧基苄基、2,4-二甲氧基苄基、邻甲基苄基、间甲基苄基、对甲基苄基、邻氯苄基、对氯苄基、间氯苄基、苯甲酰基、甲氧羰基、叔丁氧羰基或苄氧羰基;n为1、2或3。
根据本发明优选的,所述式ii化合物为n-苄基-2s-甲氧羰基甲基亚氨基二乙酸二甲酯、n-苄基-2s-乙氧羰基甲基亚氨基二乙酸二乙酯、n-苄基-2s-甲氧羰基乙基亚氨基二乙酸二甲酯、n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯、n-苄基-2r-甲氧羰基乙基亚氨基二乙酸二甲酯、n-苄基-2r-乙氧羰基乙基亚氨基二乙酸二乙酯、n-苄基-2s-甲氧羰基丙基亚氨基二乙酸二甲酯、n-苄基-2s-乙氧羰基丙基亚氨基二乙酸二乙酯、n-对氯苄基-2s-异丙氧羰基乙基亚氨基二乙酸二异丙基酯或n-苄氧羰基-2s-乙氧羰基甲基亚氨基二乙酸二乙酯。
根据本发明优选的,式ii化合物的环化反应是于溶剂中、环化试剂的作用下进行的。
优选的,所述溶剂为四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、甲基叔丁基醚、甲氧基环戊烷、二氯甲烷、氯仿、1,2-二氯乙烷、正己烷、正庚烷或甲苯之一或组合;所述溶剂与式ⅱ化合物的质量比为(3-10):1。
优选的,所述环化试剂为路易斯酸和路易斯碱的配合物;所述路易斯酸为三氯化铝、三氟化硼、四氯化钛或四氯化锡,所述路易斯碱为三甲胺、三乙胺、三正丁胺、二异丙基乙胺或吡啶之一或组合;所述路易斯酸、路易斯碱和式ⅱ化合物的摩尔比为(1.0-4.0):(1.0-4.0):1。
优选的,所述环化反应温度为-60-50℃;进一步优选的,所述环化反应温度为-30-20℃;最优选的,所述环化反应温度为0-15℃。所述环化反应时间为0.5-5小时;进一步优选的,所述环化反应时间为1-3小时。环化反应温度过高,会使原料发生聚合副反应,从而产生副产物,降低目标产物的光学纯度以及收率。
根据本发明优选的,式ii化合物经环化反应后,不经分离直接进行下一步骤。
根据本发明优选的,所述脱羧反应是于n,n-二甲基甲酰胺(dmf)、氯化锂存在下经热脱羧反应制备得到式ⅰ化合物,或在酸存在下经水解脱羧反应制备式ⅰ化合物的盐。
优选的,所述dmf和式ⅱ化合物的质量比为(2.0-10.0):1,所述氯化锂的质量是式ⅱ化合物质量的2.0-20.0%;进一步优选的,所述dmf和式ⅱ化合物的质量比为(2.0-5.0):1,所述氯化锂的质量是式ⅱ化合物质量的5.0-10.0%。
优选的,所述热脱羧反应温度为100-180℃;进一步优选的,所述热脱羧反应温度为130-150℃。所述热脱羧反应时间为1-10小时;进一步优选的,所述热脱羧反应时间为2-5小时。热脱羧反应温度过高热分解副产物较多。
优选的,所述酸为盐酸、氢溴酸、氢碘酸、乙酸、硫酸或磷酸中的一种或两种以上的组合;进一步优选的,所述酸为盐酸;所述酸和式ⅱ化合物的摩尔比为(1.0-10.0):1;进一步优选的,所述酸和式ⅱ化合物的摩尔比为(4.0-10.0):1。
优选的,所述水解脱羧反应温度为30-110℃;进一步优选的,所述水解脱羧反应温度为60-100℃;最优选的,所述水解脱羧反应温度为60-80℃。所述水解脱羧反应时间为1-10小时;进一步优选的,所述水解脱羧反应时间为1-5小时。反应温度过高副产物较多。
根据本发明优选的,式ⅰ化合物的盐是式ⅰ化合物的盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、磷酸盐或乙酸盐中的一种。
本发明的反应路线如下所示:
其中,式ii化合物结构式中,取代基r1为甲基、乙基、正丙基、异丙基、叔丁基、正丁基、仲丁基或苄基;取代基p为苄基、邻甲氧基苄基、间甲氧基苄基、对甲氧基苄基、2,4-二甲氧基苄基、邻甲基苄基、间甲基苄基、对甲基苄基、邻氯苄基、对氯苄基、间氯苄基、苯甲酰基、甲氧羰基、叔丁氧羰基或苄氧羰基;n为1、2或3。式ii化合物经环化反应所得中间产物中,n、取代基p、取代基r1与式ii化合物结构式中的n、取代基p、取代基r1具有相同的含义;当n为1时,指:与coor1相连的双键碳直接和与取代基g相连的碳连接;取代基g为cooh或coor,其中r为甲基、乙基、正丙基、异丙基、叔丁基、正丁基、仲丁基或苄基,取代基r与式ii化合物结构式中的取代基r1具有相同的含义。式ⅰ化合物结构式中,取代基p、n、取代基g与上述中间产物中的取代基p、n、取代基g具有相同的含义。
本发明的技术特点及有益效果如下:
1、本发明提供一种高光学纯度n-p取代基-2-(r或s)-g取代基氧代氮杂环烷或其盐的制备方法,以(r或s)n-p取代基-2-烷氧羰基烷基亚氨基二乙酸二酯为原料,于溶剂和环化试剂(路易斯酸-路易斯碱)作用下进行环化反应得到n-p取代基-2-(r或s)-g取代基-(n+1)-烷氧羰基氧代氮杂环烷,不需分离,直接经热脱羧或水解脱羧反应,得到高光学纯度的n-p取代基-2-(r或s)-g取代基氧代氮杂环烷或其盐。
2、本发明的方法和现有技术相比,所用原料价廉易得,成本低,反应条件易于控制和实现;本发明所用特定种类的环化试剂利于制备高光学纯度的目标产物,且易于回收,减少了废液排放,绿色环保,有利于绿色工业化生产;所涉及的原料和产物的手性结构稳定性好,所得目标产物光学纯度和收率高。本发明所用原料(式ii化合物)的手性碳原子处于酯基的邻位,常规强碱条件下缩合时可以经由碳负离子消旋化,在本发明的反应条件下,手性碳原子保持惰性,手性保持,反应的选择性好。本发明高光学纯度n-p取代基-2-(r或s)-g取代基氧代氮杂环烷或其盐的制备为研究系列手性氧代氮杂环烷衍生物的生物活性奠定了基础。
附图说明
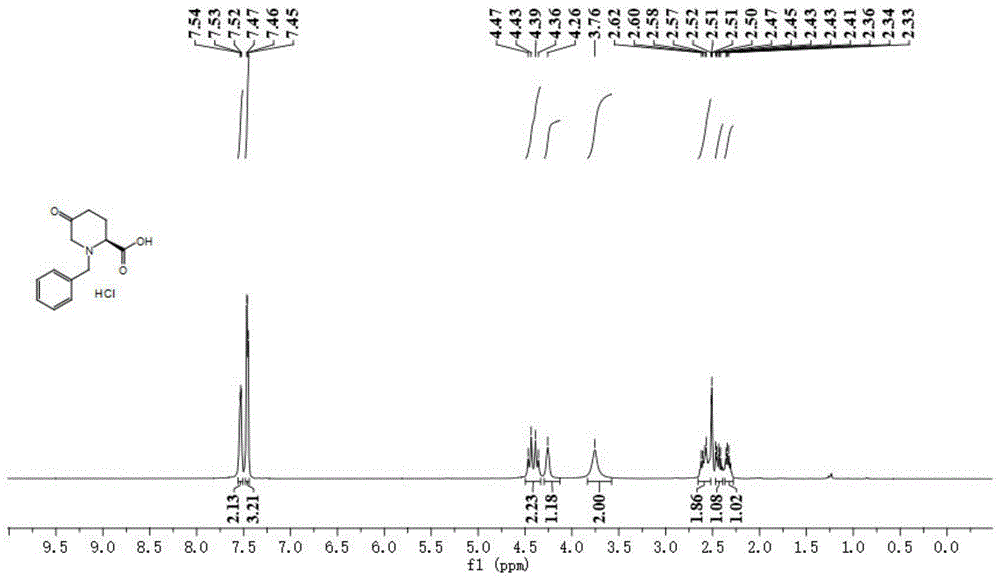
图1为本发明实施例1所得目标产物的核磁氢谱图。
图2为本发明实施例1所得目标产物的核磁碳谱图。
图3为本发明实施例1所得目标产物的正相hplc图。
图4为本发明对比例1所得产物的正相hplc图。
具体实施方式
以下结合实施例详细说明了本发明,但本发明不仅局限于此。
实施例所用原料(r或s)n-p取代基-2-烷氧羰基烷基亚氨基二乙酸二酯可按现有技术制备得到,其余原料和试剂均为市售品。
实施例中的%均为质量百分比,有特别说明的除外。
利用配有手性柱(es-ovs,150mm×4.6mm,安捷伦公司)的液相色谱仪监控反应过程和产品光学纯度(面积比%),并计算摩尔收率和ee.%值。
实施例1:(s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ1)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入100克四氢呋喃,100克二氯甲烷,37.9克(0.10摩尔)n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯(ⅱ1),40.0克(0.3摩尔)三氯化铝,搅拌均匀,冷却至-5~0℃,并于该温度下滴加15.2克(0.15摩尔)三乙胺,1-2小时滴毕,10~15℃搅拌反应3小时,将反应液体慢慢倒至100克水中,分层,有机相用100克5wt%碳酸氢钠水溶液洗涤一次,分层,有机相浓缩至干,向浓缩物中加入100克30wt%盐酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,水相减压蒸馏得到19.5克(s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ1)。
本实施例所得目标产物的正相hplc图如图3所示,由图可知,本实施例所得目标产物的光学纯度100.0%,摩尔收率为72.3%。本实施例所得目标产物的比旋度为:[α]20d=-26.2°(c=0.01,水)。
本实施例所得目标产物的核磁氢谱图以及核磁碳谱图分别如图1和2所示。
所得目标产物的核磁数据如下:1hnmr(400mhz,dmso-d6)δ:7.47(m,5h),4.41(dd,j=31.2,12.9hz,2h),4.26(s,1h),3.76(s,2h),2.59(m,2h),2.44(m,1h),2.34(m,1h)。
13c-nmr(100mhz,dmso-d6)δ:200.61,169.98,131.64,130.23,130.03,129.32,58.30,57.84,35.81,23.96。
实施例2:(s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ1)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入100克四氢呋喃,100克二氯甲烷,37.9克(0.10摩尔)n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯(ⅱ1),滴加56.9克(0.30摩尔)四氯化钛,0.5-1小时滴毕,搅拌均匀,冷却至-5~0℃,并于该温度下滴加15.2克(0.15摩尔)三乙胺,1-2小时滴毕,0~5℃搅拌反应3小时,将反应液体慢慢倒至100克水中,分层,有机相用100克5wt%碳酸氢钠水溶液洗涤一次,分层,有机相浓缩至干,向浓缩物中加入100克30wt%盐酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,水相减压蒸馏得到21.2克(s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ1),光学纯度100.0%,摩尔收率为78.6%。
实施例3:(s)-1-苄基-5-氧代哌啶-2-甲酸乙酯(ⅰ2)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入100克四氢呋喃,100克二氯甲烷,37.9克(0.10摩尔)n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯(ⅱ1),40.0克(0.3摩尔)三氯化铝,搅拌均匀,冷却至-5~0℃,并于该温度下滴加15.2克(0.15摩尔)三乙胺,1-2小时滴毕,0~5℃搅拌反应3小时,将反应液体慢慢倒至100克水中,分层,有机相用100克5wt%碳酸氢钠水溶液洗涤一次,分层,有机相浓缩至干,向浓缩物中加入100克dmf,3.0克氯化锂,130~135℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,减压蒸馏(130-140℃/1-1.5mmhg)得到19.7克(s)-1-苄基-5-氧代哌啶-2-甲酸乙酯(ⅰ2),光学纯度100.0%,摩尔收率为75.5%。
实施例4:(r)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ3)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入100克四氢呋喃,100克二氯甲烷,37.9克(0.10摩尔)n-苄基-2r-乙氧羰基乙基亚氨基二乙酸二乙酯(ⅱ2),滴加56.9克(0.30摩尔)四氯化钛,1-1.5小时滴毕,搅拌均匀,冷却至-5~0℃,并于该温度下滴加15.2克(0.15摩尔)三乙胺,1-2小时滴毕,0~5℃搅拌反应3小时,将反应液体慢慢倒至100克水中,分层,有机相用100克5wt%碳酸氢钠水溶液洗涤一次,分层,有机相浓缩至干,向浓缩物中加入100克30wt%盐酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,水相减压蒸馏得到21.1克(r)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ3),光学纯度100.0%,摩尔收率为78.2%,比旋度为:[α]20d=+26.2°(c=0.01,水)。
实施例5:(s)-1-苄氧羰基-4-氧代四氢吡咯烷-2-羧酸氢溴酸盐(ⅰ4)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入350克二氯甲烷,40.9克(0.10摩尔)n-苄氧羰基-2s-乙氧羰基甲基亚氨基二乙酸二乙酯(ⅱ3),滴加56.9克(0.30摩尔)四氯化钛,0.5-1小时滴毕,冷却至-10~0℃,搅拌2小时,并于该温度下滴加30.3克(0.30摩尔)三乙胺,1-2小时滴毕,0~5℃搅拌反应3小时,加水淬灭,分层,有机相浓缩至干,向浓缩物中加入100克40wt%氢溴酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,得到23.4克(s)-1-苄氧羰基-4-氧代四氢吡咯烷-2-羧酸氢溴酸盐(ⅰ4),光学纯度100.0%,摩尔收率为78.6%,比旋度为[α]20d=18.3°(c=0.01,二氯甲烷)。
所得目标产物的核磁数据如下:
1hnmr(400mhz,dmso-d6)δ:7.47(m,5h),4.68(s,2h),3.70(m,1h,),3.43(d,j=16.8hz,1h),3.15(d,j=16.8hz,1h),2.75(m,1h),2.63(m,1h)。
实施例6:(s)-1-对氯苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ6)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入100克四氢呋喃,100克二氯甲烷,45.6克(0.10摩尔)n-对氯苄基-2s-异丙氧羰基乙基亚氨基二乙酸二异丙基酯(ⅱ4),40.0克(0.3摩尔)三氯化铝,搅拌均匀,冷却至-5~0℃,并于该温度下滴加15.2克(0.15摩尔)三乙胺,1-2小时滴毕,10~15℃搅拌反应3小时,将反应液体慢慢倒至100克水中,分层,有机相用100克5wt%碳酸氢钠水溶液洗涤一次,分层,有机相浓缩至干,向浓缩物中加入100克30wt%盐酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,水相减压蒸馏得到22.3克(s)-1-对氯苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ6),摩尔收率为73.2%。
所得目标产物的核磁数据如下:
1hnmr(400mhz,dmso-d6)δ:7.51(d,2h),7.43(d,2h),4.41(dd,j=31.2,12.9hz,2h),4.26(s,1h),3.76(s,2h),2.59(m,2h),2.44(m,1h),2.34(m,1h)。
对比例1:(r,s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ5)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入200克四氢呋喃,10.8克(0.2摩尔)甲醇钠,搅拌均匀,冷却至-5~0℃,并于该温度下滴加37.9克(0.10摩尔)n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯(ⅱ1),1-2小时滴毕,0~5℃搅拌反应3小时,将反应液体慢慢倒至100克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,有机相浓缩至干,向浓缩物中加入100克30wt%盐酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,水相减压蒸馏得到20.1克(r,s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ5)。
本对比例所得产物的正相hplc图如图4所示,由图可知,所得产物的光学纯度0.0%,摩尔收率为74.6%。所得产物的比旋度为:[α]20d=0°(c=0.01,水)。
对比例1表明n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯于强碱条件下进行环化反应时,原手性碳原子发生了消旋化,进一步表明该反应条件不适于高光学活性目标化合物的制备。
对比例2:(s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ1)的制备
向装有搅拌、温度计和恒压滴液漏斗的500毫升四口烧瓶中加入200克四氢呋喃,37.9克(0.10摩尔)n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯(ⅱ1),40.0克(0.3摩尔)三氯化铝,搅拌均匀,冷却至-5~0℃,并于该温度下滴加15.2克(0.15摩尔)三乙胺,1-2小时滴毕,55~60℃搅拌反应3小时,将反应液体慢慢倒至100克水中,分层,有机相用100克5wt%碳酸氢钠水溶液洗涤一次,分层,有机相浓缩至干,向浓缩物中加入100克30wt%盐酸,60~65℃搅拌反应2小时,冷却至20~25℃,将所得反应液体加到50克水和100克二氯甲烷混合物中,分层,水层用二氯甲烷萃取,每次50克,合并有机层,常压蒸馏回收溶剂,水相减压蒸馏得到9.8克(s)-1-苄基-5-氧代哌啶-2-羧酸盐酸盐(ⅰ1),光学纯度96.3%,收率为56.7%。
对比例2表明环化反应温度高,收率降低,分析原因为n-苄基-2s-乙氧羰基乙基亚氨基二乙酸二乙酯于高温条件下进行了聚合反应,同时产品的光学纯度低。
- 还没有人留言评论。精彩留言会获得点赞!