十七元大环脂肽天然化合物的合成方法与流程
本发明属于有机合成技术领域,尤其涉及一种十七元大环脂肽天然化合物的合成方法。
背景技术:
具有逆转肿瘤细胞的多药耐药活性的十七元大环脂肽类天然产物dysoxylactama是由岳建民院士课题组(yue,j.m.,etal.j.am.chem.soc.2019,141,6812.)在2019年从香港樫木(dysoxylumhongkongense)的树皮中分离得到。从结构上分析,dysoxylactama是具有6个手性中心的十七元大环缩酯肽,同时侧链是一个独特的含有五个连续立体中心的脂肪酸。初步的生物活性研究表明,dysoxylactama能够逆转肿瘤细胞的多药耐药现象。通过体外细胞实验发现,由阿霉素(adriamycin)、长春新碱(vincristine)和紫杉醇(paclitaxel)等化学药引发多药耐药的肿瘤细胞,经过低浓度的dysoxylactama处理之后,对这些抗癌药的响应提升了28.4-1039.7倍,更重要的是,在10μm的浓度下,dysoxylactama并没有体现任何细胞毒性。说明dysoxylactama显著恢复了p-gp(p-糖蛋白)底物药物(如阿霉素,长春新碱和紫杉醇)对这些耐药性癌细胞的敏感性,并且无明显影响关于这些药物对亲代对应细胞的敏感性。逆转癌细胞多药耐药的抑制剂通常通过两种方式起效:一种是抑制p-gp的表达以降低其数量,从而降低癌细胞释放抗癌药物的可能性;另一种是直接与p-gp结合,以降低其转运功能。对肿瘤细胞mcf7/adr的研究发现,dysoxylactama并未显著降低p-gp的表达,而是抑制肿瘤细胞中p-gp的转运功能。
鉴于dysoxylactama具有优异的逆转肿瘤细胞多药耐药现象的生物活性,它有望成为治疗癌症多药耐药的突破口。在完成活性天然产物dysoxylactama的合成的基础上,进一步修饰并优化天然产物的结构,探索其构效关系,并探寻它与靶标蛋白p-gp的作用模式,尝试阐明dysoxylactama逆转肿瘤细胞多药耐药的作用机制。
技术实现要素:
本发明的目的在于提供一种十七元大环脂肽天然化合物的合成方法,旨在解决现有dysoxylactama只能从香港樫木的树皮中分离提取,提取效率低,且无法实现在dysoxylactama的基础上进一步修饰化合物结构,探索构效关系等技术问题。
为了实现上述发明目的,本发明采用的技术方案如下:
一种十七元大环脂肽天然化合物的合成方法,其特征在于,包括一下步骤:
获取(r)-3-羟基异丁酸甲酯,对所述(r)-3-羟基异丁酸甲酯依次进行还原缩合反应、锂化-硼基化反应、迁移反应、锂卤交换反应和氧化处理,得到化合物9;
对所述化合物9进行不对称布朗丁烯基化反应,得到化合物3;
将所述化合物3与氨基苄氧羰基保护的左旋缬氨酸通过酯化反应,得到化合物12;
对所述化合物12进行催化氢化反应后与6-庚烯酸进行接肽反应,得到化合物14;
对所述化合物14进行烯丙基化反应,得到化合物2;
对所述化合物2进行关环复分解反应,得到化合物15;
对所述化合物15进行催化氢化反应,得到dysoxylactama;
其中,所述化合物9为:
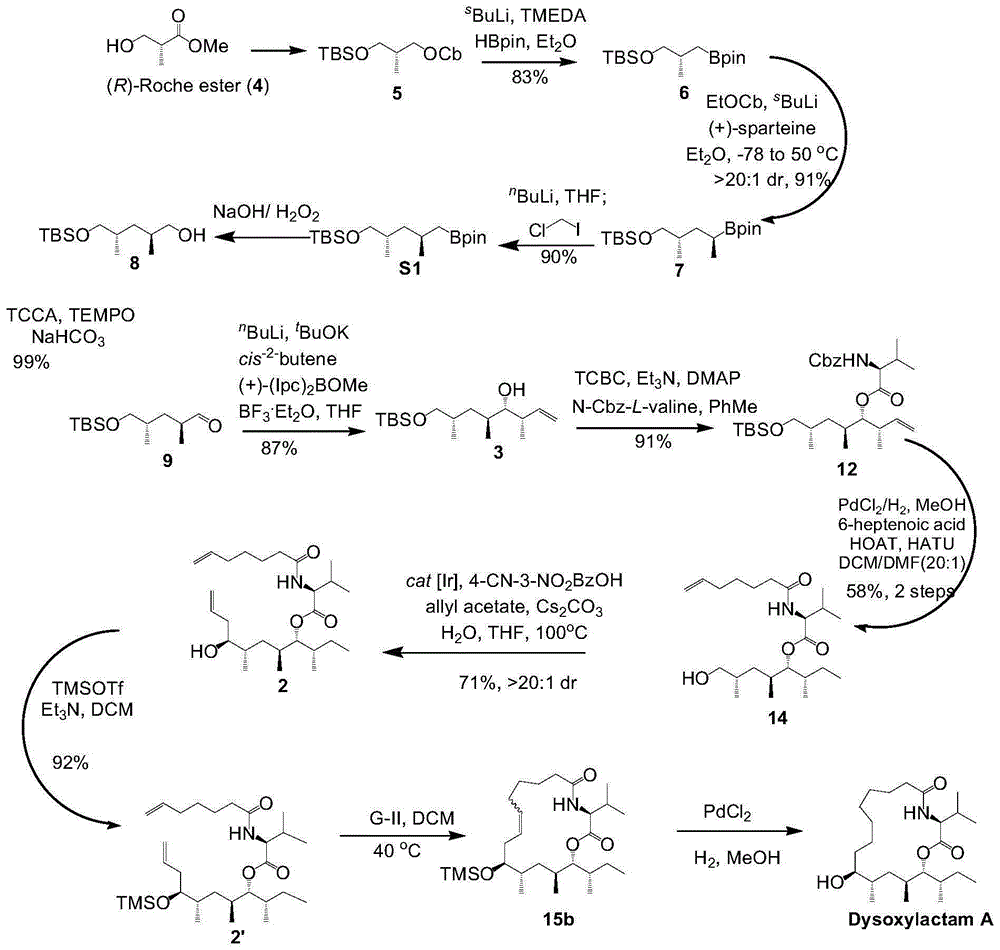
本发明提供的十七元大环脂肽天然化合物的合成方法,通过对目标产物dysoxylactama的结构进行分析设计了上述逆合成路线,具体地:dysoxylactama结构中含有的酯键和酰胺键是大环化合物常用的关环位点,但都存在一定位阻,本申请拟采用烯烃复分解反应(rcm)实现大环的构建,切断c6-c7碳碳键可以逆推至前体化合物2。线性关环前体化合物2可以通过酰胺化(amidation)和酯化反应(yamaguchiesterification)分别引入所需的6-庚烯酸和l-缬氨酸。c9位的手性羟基可以通过手性ir催化剂催化的krischeallylation(krische烯丙基化反应)高立体选择性的构建。故此,通过以上策略可以将这一复杂的前体化合物2简化至tbs单保护的二醇化合物3,分子中c13位的手性羟基可以使用browncrotylation(布朗丁烯基化反应)高选择性的构建,同时引入含有所需手性中心的巴豆基,再利用的锂化-硼基化技术(aggarwalhomologation)快速进行碳碳键组装等一系列转化可以逆推到商业可得的化合物(r)-3-羟基异丁酸甲酯(r-rocheester),化合物4。本发明通过逆推导过程设计的上述合成路线,以(r)-3-羟基异丁酸甲酯为原料,依次经过还原缩合反应、锂化-硼基化反应、迁移反应、锂卤交换反应、氧化处理、不对称布朗丁烯基化反应、酯化反应、接肽反应、烯丙基化反应、关环复分解反应以及催化氢化反应,合成得到十七元大环脂肽天然化合物dysoxylactama,合成产率高,产物药用价值高。
附图说明
图1是本发明实施例提供的dysoxylactama的合成路线示意图。
具体实施方式
为使本发明实施例的目的、技术方案和技术效果更加清楚,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。结合本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
在本发明的描述中,需要理解的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
本发明实施例说明书中所提到的相关成分的重量不仅仅可以指代各组分的具体含量,也可以表示各组分间重量的比例关系,因此,只要是按照本发明实施例说明书相关组分的含量按比例放大或缩小均在本发明实施例说明书公开的范围之内。具体地,本发明实施例说明书中所述的重量可以是μg、mg、g、kg等化工领域公知的质量单位。
本发明实施例提供了一种十七元大环脂肽天然化合物的合成方法,包括以下步骤:
s00.获取(r)-3-羟基异丁酸甲酯,对所述(r)-3-羟基异丁酸甲酯依次进行还原缩合反应、锂化-硼基化反应、迁移反应、锂卤交换反应和氧化处理,得到化合物9;
s60.对所述化合物9进行不对称布朗丁烯基化反应,得到化合物3;
s70.将所述化合物3与氨基苄氧羰基保护的左旋缬氨酸通过酯化反应,得到化合物12;
s80.对所述化合物12进行催化氢化反应后与6-庚烯酸进行接肽反应,得到化合物14;
s90.对所述化合物14进行烯丙基化反应,得到化合物2;
s11.对所述化合物2进行关环复分解反应,得到化合物15;
s12.对所述化合物15进行催化氢化反应,得到dysoxylactama;
其中,所述化合物9为:
本发明实施例提供的十七元大环脂肽天然化合物的合成方法,通过对目标产物dysoxylactama的结构进行分析设计了上述逆合成路线,具体地:dysoxylactama结构中含有的酯键和酰胺键是大环化合物常用的关环位点,但都存在一定位阻,本申请拟采用烯烃复分解反应(rcm)实现大环的构建,切断c6-c7碳碳键可以逆推至前体化合物2。线性关环前体化合物2可以通过酰胺化(amidation)和酯化反应(yamaguchiesterification)分别引入所需的6-庚烯酸和l-缬氨酸。c9位的手性羟基可以通过手性ir催化剂催化的krischeallylation(krische烯丙基化反应)高立体选择性的构建。故此,通过以上策略可以将这一复杂的前体化合物2简化至tbs单保护的二醇化合物3,分子中c13位的手性羟基可以使用browncrotylation(布朗丁烯基化反应)高选择性的构建,同时引入含有所需手性中心的巴豆基,再利用的锂化-硼基化技术(aggarwalhomologation)快速进行碳碳键组装等一系列转化可以逆推到商业可得的化合物(r)-3-羟基异丁酸甲酯(r-rocheester),化合物4,推导过程如下所示。本发明实施例通过逆推导过程设计的上述合成路线,以(r)-3-羟基异丁酸甲酯为原料,依次经过还原缩合反应、锂化-硼基化反应、迁移反应、锂卤交换反应、氧化处理、不对称布朗丁烯基化反应、酯化反应、接肽反应、烯丙基化反应、关环复分解反应以及催化氢化反应,合成得到十七元大环脂肽天然化合物dysoxylactama,合成产率高,产物药用价值高。
具体地,上述步骤s00中获取(r)-3-羟基异丁酸甲酯,对所述(r)-3-羟基异丁酸甲酯依次进行还原缩合反应、锂化-硼基化反应、迁移反应、锂卤交换反应和氧化处理的步骤包括:
s10.获取(r)-3-羟基异丁酸甲酯,对所述(r)-3-羟基异丁酸甲酯进行还原缩合反应,得到化合物5,
s20.对所述化合物5进行锂化-硼基化反应,得到化合物6,
s30.对所述化合物6进行迁移反应,得到化合物7,
s40.对所述化合物7进行锂卤交换反应,得到化合物8,
s50.对所述化合物8进行氧化处理,得到化合物9。
本发明实施例从商业已知的从(r)-3-羟基异丁酸甲酯合成制备化合物3的过程中,首先,以商业已知的从(r)-3-羟基异丁酸甲酯为起始原料,在三乙胺和叔丁基二甲基氯硅烷(tbscl)的作用下将一级羟基用tbs保护。然后,在室温下,使用二异丁基氢化铝将分子中的甲酯还原为羟基,然后和二异丙基甲胺酰氯(cbcl)通过酯缩合得到已知化合物5。然后,通过锂化-硼基化反应使得ocb基团离去,而引入bpin基团(嚬哪醇硼酸酯),形成有机硼试剂化合物6。然后,通过仲丁基锂拔去碳酸酯α位的氢在右旋金鹰爪碱的络合作用下形成手性锂试剂,和化合物6形成硼盐,再通过1,2-迁移以很好的立体选择性完成手性甲基的构建,得化合物7。然后,在氯碘甲烷和正丁基锂的条件下通过锂卤交换延长碳链,紧接着反应液直接用氢氧化钠和过氧化氢溶液处理即可得到羟基化合物8。然后,化合物8在碳酸氢钠存在的情况下发生四甲基六氢吡啶氧化物氧化(tempo),氧化成醛化合物9。最后,化合物9通过不对称布朗丁烯基化(browncrotylation)可以高度立体选择性的实现两个连续手性中心的构建,完成tbs单保护的二醇化合物3的制备。在一些具体实施例中,从(r)-3-羟基异丁酸甲酯合成制备化合物3的制备过程如下所示:
具体地,上述步骤s10中,对所述(r)-3-羟基异丁酸甲酯进行还原缩合反应的步骤包括:采用tbs对所述(r)-3-羟基异丁酸甲酯的羟基进行保护后,在温度为25℃~38℃的条件下,依次添加二异丁基氢化铝进行还原反应和二异丙基甲胺酰氯进行缩合反应,分离得到化合物5。在一些实施例中,所述(r)-3-羟基异丁酸甲酯、所述二异丁基氢化铝和所述二异丙基甲胺酰氯的摩尔比为(0.5~1.5):(2.5~3.5):(3.5~4.5)。本发明通过实施例配比的各原料组分,以(r)-3-羟基异丁酸甲酯为起始原料,在三乙胺和叔丁基二甲基氯硅烷的作用下将一级羟基用tbs保护;然后在25℃~38℃的室温下,采用二异丁基氢化铝将分子中的甲酯还原为羟基后,与cbcl通过酯缩合得到化合物5,各原料组分的配比有效确保了原料物质间反应充分,减少副产物生成,化合物5的产率高,纯度好。
具体地,上述步骤s20中,对所述化合物5进行锂化-硼基化反应的步骤包括:在温度为-40℃~-80℃的条件下,将仲丁基锂添加到所述化合物5和四甲基乙二胺的混合溶液中反应5~8小时,添加频哪醇硼烷反应1~3小时后,升温至30~50℃反应10~15小时,分离得到化合物6。在一些实施例中,所述仲丁基锂、所述化合物5、所述四甲基乙二胺和所述频哪醇硼烷的摩尔比为(2~3):(1~12):(2.5~3.5):(3.5~4.5)。本发明实施例在上述物质配比的情况下,通过锂化-硼基化反应离去ocb基团,而引入嚬哪醇硼酸酯(bpin基团),得到有机硼试剂化合物6,各原料组分的配比有效确保了原料物质间反应充分,减少副产物生成。
在一些实施例中,所述化合物5和四甲基乙二胺的混合溶液中溶剂选自:无水乙醚、四氢呋喃、甲基叔丁基醚中的至少一种,这些溶剂对化合物5、四甲基乙二胺、频哪醇硼烷等物质组分均有较好的溶解性。
在一些具体实施例中,在-40℃~-80℃将sbuli缓慢滴加到氨基甲酸酯化合物5和tmeda的无水乙醚溶液中,在-40℃~-80℃反应5~8个小时然后缓慢滴加hbpin的乙醚溶液中。在-40℃~-80℃反应1~3个小时后撤去冷阱,反应液升温至30℃~50℃继续反应10~15个小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入kh2po4水溶液淬灭反应,继续搅拌10~30分钟。反应液用乙醚萃取,合并得到的有机相先后用水和饱和食盐水洗涤洗涤,再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状有机硼试剂化合物6。
具体地,上述步骤s30中,对所述化合物6进行迁移反应的步骤包括:在温度为-40℃~-80℃的条件下,将仲丁基锂添加到乙基氨基甲酸酯和(+)-金鹰爪碱的混合溶液中反应3~5小时,添加化合物6反应1~3小时后,升温至30~50℃反应10~15小时,分离得到化合物7。在一些实施例中,所述仲丁基锂、所述乙基氨基甲酸酯、所述(+)-金鹰爪碱和所述化合物6的摩尔比为(1~2):(0.5~1.5):(1~2):(0.2~0.5)。本发明实施例通过仲丁基锂拔去碳酸酯α位的氢后在右旋金鹰爪碱的络合作用下形成手性锂试剂,然后和化合物6形成硼盐再通过1,2-迁移以很好的立体选择性完成手性甲基的构建,得到化合物7。各原料组分的配比有效确保了原料物质间反应充分,减少副产物生成。
在一些实施例中,所述乙基氨基甲酸酯和(+)-金鹰爪碱的混合溶液中溶剂选自:无水乙醚、四氢呋喃、甲基叔丁基醚中的至少一种。
在一些具体实施例中,在-40℃~-80℃将sbuli(仲丁基锂)缓慢滴加到乙基氨基甲酸酯和(+)-金鹰爪碱((+)-sparteine)的无水乙醚溶液中,在-40℃~-80℃反应3~5个小时然后缓慢滴加化合物6的乙醚溶液中。在-40℃~-80℃反应1~3个小时后撤去冷阱,反应液升温至30~50℃继续反应10~15个小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入kh2po4水溶液淬灭反应,继续搅拌10分钟。反应液用乙醚萃取,合并得到的有机相先后用水和饱和食盐水洗涤洗涤,再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状有机硼酸酯化合物7。
具体地,上述步骤s40中,对所述化合物7进行锂卤交换反应:在温度为-40℃~-80℃的条件下,将正丁基锂添加到所述化合物7与碘氯甲烷的混合溶液后,升温至25℃~38℃反应10~15小时,分离得到化合物8。在一些实施例中,所述化合物7、所述碘氯甲烷和正丁基锂的摩尔比为(0.2~0.5):(3~8):(1~2)。本发明实施例按所述化合物7、所述碘氯甲烷和正丁基锂的摩尔比为(0.2~0.5):(3~8):(1~2),使化合物7在氯碘甲烷和正丁基锂的条件下通过锂卤交换延长碳链,紧接着反应液直接用氢氧化钠和过氧化氢溶液处理,即可得到羟基化合物8。各原料组分的配比有效确保了原料物质间反应充分,减少副产物生成。
在一些实施例中,所述化合物7与碘氯甲烷的混合溶液中的溶剂选自:四氢呋喃、乙醚、甲基叔丁基醚中的至少一种。
在一些具体实施例中,将化合物7和碘氯甲烷溶于极其干燥的thf中,-40℃~-80℃条件下缓慢滴加正丁基锂溶液,(如果滴加速度过快溶液会变成黄色,生成大量副产物)。随后将反应体系升至室温并持续搅拌10~15小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入2nnaoh/30%wth2o2水溶液淬灭反应,继续搅拌30~50分钟。反应液用乙醚萃取,合并得到的有机相先后用水和饱和食盐水洗涤,再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离,即可得到无色油状醇化合物8。
在另一些实施例中,将化合物7和碘氯甲烷溶于极其干燥的thf中,-40℃~-80℃条件下缓慢滴加正丁基锂溶液,随后将反应体系升至室温并持续搅拌10~15小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入kh2po4水溶液淬灭反应,继续搅拌30~50分钟。反应液用乙醚萃取,合并得到的有机相先后用水和饱和食盐水洗涤洗涤,再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状化合物s1中间体,
具体地,上述步骤s50中,对所述化合物8进行氧化处理的步骤包括:将碳酸氢钠、三氯异氰脲酸和四甲基哌啶氮氧化物添加到所述化合物8的溶液中,在温度为0℃~5℃的条件下反应2~5分钟,分离得到化合物9。在一些实施例中,所述化合物8、所述碳酸氢钠、所述三氯异氰脲酸和所述四甲基哌啶氮氧化物的摩尔比为(0.3~1):(1.5~2.5):(0.5~1):(0.03~0.1)。本发明实施例化合物8在碳酸氢钠存在的情况下发生tempo氧化反应,氧化成醛化合物9。各原料组分的配比有效确保了原料物质间反应充分,减少副产物生成。
在一些实施例中,所述化合物8的溶液中溶剂选自:二氯甲烷、甲苯、乙腈中的至少一种。
在一些具体实施例中,将化合物8溶解于二氯甲烷中,冷却至0℃~5℃后依次加入nahco3,tcca(三氯异氰脲酸)和tempo(四甲基哌啶氮氧化物)。反应体系在0℃~5℃条件下搅拌2~5分钟后,反应液通过硅藻土过滤然后用na2s2o3水溶液淬灭反应。反应液用dcm(二氯甲烷)萃取,合并得到的有机相用饱和的nahco3水溶液,再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离,即可得到无色油状化合物9。
具体地,上述步骤s60中,对所述化合物9进行不对称布朗丁烯基化反应的步骤包括:在温度为-80℃~-30℃的无水无氧环境下,将所述化合物9与叔丁醇钾、顺式2-丁烯、正丁基锂、(+)-ipc2bome和三氟化硼乙醚溶解于第一有机溶剂中后反应10~15小时,分离得到化合物3。在一些实施例中,所述化合物9、所述叔丁醇钾、所述顺式2-丁烯、所述正丁基锂、所述(+)-ipc2bome和所述三氟化硼乙醚的摩尔比为(1~1.5):(2.2~2.8):(6~6.5):(2.2~2.8):(3.5~4):(4.8~5.3)。本发明实施例将化合物9通过不对称browncrotylation反应(布朗丁烯基化反应)可以高度立体选择性的实现两个连续手性中心的构建,完成tbs单保护的二醇化合物3的制备。各原料组分的配比有效确保了原料物质间反应充分,减少副产物生成。
在进一步实施例中,对所述化合物9进行不对称布朗丁烯基化反应的步骤包括:在无氧环境中,将干燥无氧的所述叔丁醇钾溶解在第一有机溶液后冷却至-60℃~-80℃,依次添加所述顺式2-丁烯和所述正丁基锂,升温至-30℃~-45℃反应10~30分钟;然后降温到-60℃~-80℃添加所述(+)-ipc2bome反应10~30分钟,再依次添加所述三氟化硼乙醚和所述化合物9,在温度为-60℃~-80℃的条件下反应10~15小时。
在一些实施例中,所述第一有机溶剂选自:四氢呋喃、乙醚、甲基叔丁基醚中的至少一种。
在一些具体实施例中,称取t-buok(叔丁醇钾)于烧瓶中在80~100℃下抽真空过夜,然后加入thf将其溶解,冷却到-60℃~-80℃加入顺式2-丁烯,随后逐滴滴加n-buli,伴随着反应液变成橘黄色。然后升温至-30℃~-45℃反应10~30分钟然后再降温到-60℃~-80℃。紧接着在手套箱中称取(+)-ipc2bome并溶解在thf加入,逐滴滴加同时反应液变成亮黑色。10~30分钟后逐滴滴加bf3·oet2(三氟化硼乙醚),紧接着将化合物9溶解在thf中逐滴加入,在-60℃~-80℃反应10~15个小时,经tlc(薄层色谱)监测确认化合物9转化完全后,用naoh将反应淬灭再加入过氧化氢在50~70℃回流1~2h搅拌至澄清,用乙醚萃取合并有机相之后用饱和nh4cl溶液洗涤,无水硫酸钠固体干燥。过滤,减压蒸馏除去有机溶剂,经快速柱层析分离得到无色油状产品化合物3。
具体地,上述步骤s70中,将所述化合物3与氨基苄氧羰基保护的左旋缬氨酸通过酯化反应的步骤包括:将化合物3与氨基苄氧羰基保护的左旋缬氨酸溶解于第二有机溶剂中,在-5℃~0℃的条件下,依次添加三乙胺和2,4,6-三氯苯甲酰氯后,升温至25℃~38℃反应10~30分钟;然后降温至-5℃~0℃,添加4-二甲氨基吡啶,升温至25℃~38℃反应10~12小时,分离得到化合物12。本发明实施例制得tbs单保护的二醇化合物3后,与氨基苄氧羰基保护的左旋缬氨酸在yamaguchi酯化的条件下进行接酯反应,即可成功得到化合物12。
另外,本发明实施例还尝试了,在丙烯酸乙酯溶液中以pdcl2和氢气气氛的条件,发生催化氢化反应将双键还原后,再在yamaguchi酯化的条件下尝试相应的醇与酸化合物10进行接酯,
在一些实施例中,所述化合物3、所述氨基苄氧羰基保护的左旋缬氨酸、所述三乙胺、所述2,4,6-三氯苯甲酰氯和所述4-二甲氨基吡啶的摩尔比为(0.8~1.5):(1.8~2.5):(4~5):(3~4):(4~6),配比有效确保了原料物质间反应充分,减少副产物生成,提高目标产物纯度。
在一些实施例中,所述第二有机溶剂选自:甲苯、二氯甲烷,氮氮二甲基甲酰胺,四氢呋喃中的至少一种。
在一些具体实施例中,将化合物3和氨基苄氧羰基保护的l-缬氨酸(溶解于甲苯,在零度的条件下依次加入三乙胺和2,4,6-三氯苯甲酰氯(tcbc),然后反应液升温至室温继续搅拌10~30分钟后在零度条件下加入4-二甲氨基吡啶(dmap)。然后将反应液升温至室温反应10~12小时后经tlc(薄层色谱)监测确认转化完全后用饱和nh4cl水溶液将反应淬灭,然后用乙酸乙酯萃取合并有机相之后用饱和食盐水溶液洗涤,无水硫酸钠固体干燥。过滤,减压蒸馏除去有机溶剂,经快速柱层析分离,得到无色油状产品化合物12。
具体地,上述步骤s80中,对所述化合物12进行催化氢化反应后与6-庚烯酸进行接肽反应的步骤包括:在氮气、氩气、氦气等保护气体氛围下,将所述化合物12和第一钯催化剂溶解于第三有机溶剂后,在还原气体(氢气)氛围下反应3~5小时,得到氨基醇粗产物;然后将所述氨基醇粗产物与6-庚烯酸溶解于第四有机溶剂中,在-5℃~0℃的条件下,依次添加n,n-二异丙基乙胺、1-羟基-7-偶氮苯并三氮唑和2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯后,在25℃~38℃条件下反应8~10小时,分离得到化合物14。本发明实施例化合物12溶液在pdcl2和氢气气氛的条件中,可以发生催化氢化反应将双键还原以及脱除保护基cbz(苄氧羰基),同时由于氢化反应生成的hcl使溶液变成弱酸性环境可以脱除一级tbs保护基,因此得到氨基醇化合物13,
在一些实施例中,所述化合物12、所述第一钯催化剂、所述6-庚烯酸、所述n,n-二异丙基乙胺、所述1-羟基-7-偶氮苯并三氮唑和所述2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯的摩尔比为:(0.3~0.4):(0.1~0.2):(0.5~1):(3~4):(0.5~1):(1~2),该配比的各原料组分有效确保了原料物质间反应充分,减少副产物生成,提高目标产物纯度。
在一些实施例中,所述第一钯催化剂选自二氯化钯、钯碳、氢氧化钯碳中的至少一种。本发明实施例采用的这些钯催化剂在还原气体氛围下均能够将双键还原并脱除保护基cbz(苄氧羰基)。
在一些实施例中,所述第三有机溶剂选自:甲醇、乙酸乙酯、二氯甲烷、氯仿中的至少一种。
在一些实施例中,所述第四有机溶剂选自:体积比为(15~25):1的二氯甲烷和二甲基甲酰胺的混合溶剂。本发明实施例采用体积比为(15~25):1的二氯甲烷和二甲基甲酰胺的混合溶剂作为第四有机溶剂,较好地避免羟基接酯的情况发生进一步提高反应效率,提高反应纯度。
在一些具体实施例中,将化合物12溶解于甲醇溶液中,在室温氮气保护下加入二氯化钯,随后将反应体系中的氮气置换成氢气。在室温下继续搅拌3~5小时(通过lc-ms检测到所有的保护基被脱除,双键全部被还原),将反应通过硅藻土过滤,滤液经旋转蒸发仪减压浓缩得到黄色油状氨基醇化合物13。不用进一步纯化直接用于下一步反应。将粗产品氨基醇和6-庚烯酸溶解于dcm和dmf混合溶液中,然后在零度条件下依次加入n,n-二异丙基乙胺、1-羟基-7-偶氮苯并三氮唑和2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯,反应液升温至室温继续反应8~10个小时经tlc监测确认转化完全后用饱和nh4cl水溶液将反应淬灭,然后用dcm萃取合并有机相之后用无水硫酸钠固体干燥,过滤,减压蒸馏除去有机溶剂,经快速柱层析分离,得到无色油状产品化合物14。
具体地,上述步骤s90中,对所述化合物14进行烯丙基化反应的步骤包括:在无氧环境下,将所述化合物14、铱催化剂、4-氯-3-硝基苯甲酸和碳酸铯溶解于四氢呋喃、水和乙酸烯丙酯的混合溶液中,在温度为80~100℃的条件下反应48~72小时,分离得到化合物2。本发明实施例一开始选择将一级羟基通过dess-matin氧化反应(戴斯-马丁氧化反应),将其氧化成醛后再通过brownallylation立体选择性地建立手性羟基,但是将醇氧化醛的过程中后处理要极其小心,否则容易发生消旋。另外brownallylation操作麻烦,后处理产生的ipcoh的处理也比较繁琐。所以本发明实施例采用krischeallylation来替换brownallylation,在铱催化剂、krische的条件下进行烯丙基化反应,得到化合物2。
在一些实施例中,所述化合物14、所述铱催化剂、所述4-氯-3-硝基苯甲酸和所述碳酸铯的摩尔比为(1~2):(0.5~1):(1~2):(1.5~3),该配比的各原料组分有效确保了原料物质间反应充分,减少副产物生成,提高目标产物纯度。
在一些实施例中,所述混合溶液中,所述四氢呋喃、所述水和所述乙酸烯丙酯的体积比为(4~6)ml:(8~12)ul:(0.15~0.25)ml,其中,水和所述乙酸烯丙酯参与烯丙基化反应。
在一些具体实施例中,在氮气保护下,将化合物14,铱催化剂,4-氯-3-硝基苯甲酸和碳酸铯加入到封管中,然后通过氮气置换加入四氢呋喃溶剂紧接着加入蒸馏水和乙酸烯丙酯。将封管封严实避免空气的接触然后加热到80~100℃反应48~72小时。经tlc监测确认转化完全后反应体系降温至室温然后用二氯甲烷稀释,固体物质通过硅藻土过滤,滤液减压蒸馏出去有机溶剂得到黄色油状物质。将其吸附在硅胶上通过快速柱层析分离,得到化合物2。
其中,铱催化剂的结构式为
具体地,在一些实施例中,上述步骤s11中,对所述化合物2进行关环复分解反应的步骤包括:在无氧环境下,将所述化合物2溶解在第六有机溶剂后添加格拉布斯二代催化剂,在温度为30~40℃的条件下反应20~25小时,分离得到化合物15a,
具体地,在另一些实施例中,上述步骤s11中,对所述化合物2进行关环复分解反应的步骤包括:在-5℃~0℃的条件下,将三乙胺和三氟甲磺酸三甲基硅酯依次添加到所述化合物2的溶液中,升温至25℃~38℃反应3~5小时,分离得到化合物2’,
本发明实施例拿到关环前体后进行rcm关大环,一开始选择了羟基裸漏的底物,使用hg-ii催化剂(哈威达-格拉布斯二代催化剂)和排气处理的甲苯做溶剂,原料被消耗完但是产物化合物15a的分离产率只有13%。换用排气处理的dcm做溶剂产率有着微弱的提升达到22%,而当换用g-ii催化剂(格拉布斯二代催化剂)时产率可以达到40%,但是产率仍然不是很理想。在选择羟基保护基的时候,同时考虑到脱除羟基保护基和氢化还原新生成的双键可以一步发生从而提高路线效率,因此选用了较小的保护基tms将羟基保护,然后在g-ii催化剂的作用下以较好的产率70%获得天然产物前体化合物15b。
在一些实施例中,所述化合物2、所述三乙胺和所述三氟甲磺酸三甲基硅酯的摩尔比为(0.02~0.08):(1~1.5):(0.2~0.8)。在一些实施例中,所述化合物2或所述化合物2’和所述格拉布斯二代催化剂的摩尔比为(1~3):(0.1~0.3)。本发明上述各实施例配比的各原料组分有效确保了原料物质间反应充分,减少副产物生成,提高目标产物纯度。
在一些实施例中,所述化合物2的溶液中溶剂选自:二氯甲烷、甲苯、乙腈中的至少一种。
在一些实施例中,所述第六有机溶剂和所述第七有机溶剂分别独立地选自:二氯甲烷、甲苯、乙腈中的至少一种。
在一些具体实施例中,将化合物2溶于二氯甲烷并在零度条件下依次滴加三乙胺和tmsotf,然后反应液在室温条件下反应3~5个小时,经tlc监测确认转化完全后反应体系用饱和氯化铵溶液淬灭反应。反应液恢复至室温后用dcm萃取并合并有机相,有机相用无水硫酸钠干燥,过滤并减压蒸馏除去低沸点有机溶剂组分,所得粗产物用硅胶柱层析(乙酸乙酯/正己烷=1:4),制得无色油状化合物2′。然后,将二烯化合物2′称取在一个充分干燥的圆底烧瓶中然后安装上回流管通过氮气进行三次置换,加入提前进行除气干燥的二氯甲烷,紧接着加入格拉布斯二代催化剂溶解在提前进行除气干燥的二氯甲烷的溶液。然后将圆底烧瓶浸没在30~40℃的油浴锅中,并在此温度下反应20~25小时。经tlc监测确认转化完全后反应体系暴露在空气中搅拌10~30分钟然后通过旋转蒸发仪除去低沸点的有机溶剂,得到深棕色的油状物质化合物15b。
具体地,上述步骤s12中,对所述化合物15进行催化氢化反应的步骤包括:在保护气体氛围下,将所述化合物15和第二钯催化剂溶解于第八有机溶剂后,在还原气体氛围下反应3~4小时,分离得到dysoxylactama。本发明实施例大环化合物15b之后在甲醇溶液中以pdcl2和氢气气氛的条件,可以发生催化氢化反应将双键还原以及氢化反应生成的hcl使溶液变成弱酸性环境可以脱除二级tms保护基从而拿到最终的天然产物dysoxylactama。
在一些实施例中,所述化合物15和所述第二钯催化剂的摩尔比为(1~3):(0.01~0.02)。在一些实施例中,所述第二钯催化剂选自二氯化钯、钯碳、氢氧化钯碳中的至少一种。在一些实施例中,所述第八有机溶剂选自:甲醇、乙酸乙酯、二氯甲烷、氯仿中的至少一种。在一些实施例中,所述保护气体选自:氮气、氩气、氦气中的至少一种。在一些实施例中,所述还原气体选自:氢气。本发明上述各实施例中采用的原料组分以及提供的气体氛围有效确保了天然产物dysoxylactama的合成产率和纯度。
在一些具体实施例中,将化合物15b溶解于甲醇溶液中,在室温氮气保护下加入二氯化钯,随后将反应体系中的氮气置换成氢气。在室温下继续搅拌3~4小时(通过lc-ms检测到tms保护基被脱除,双键全部被还原),将反应通过硅藻土过滤,滤液经旋转蒸发仪减压浓缩得到的粗产品吸附在柱层析硅胶上经硅胶柱层析(乙酸乙酯/正己烷=1:2)纯化,得到白色固体天然产物dysoxylactama。
在一些实施例中,十七元大环脂肽天然化合物的合成方法,包括步骤:
s11.获取(r)-3-羟基异丁酸甲酯,采用tbs对所述(r)-3-羟基异丁酸甲酯的羟基进行保护后,依次添加二异丁基氢化铝进行还原反应和二异丙基甲胺酰氯进行缩合反应,分离得到化合物5;
s21.将仲丁基锂添加到所述化合物5和四甲基乙二胺的混合溶液中反应后,添加频哪醇硼烷反应,分离得到化合物6;
s31.将仲丁基锂添加到乙基氨基甲酸酯和(+)-金鹰爪碱的混合溶液中反应后,添加化合物6反应,分离得到化合物7;
s41.将正丁基锂添加到所述化合物7与碘氯甲烷的混合溶液反应,分离得到化合物8;
s51.将碳酸氢钠、三氯异氰脲酸和四甲基哌啶氮氧化物添加到所述化合物8的溶液中反应,分离得到化合物9;
s61.将所述化合物9与叔丁醇钾、顺式2-丁烯、正丁基锂、(+)-ipc2bome和三氟化硼乙醚溶解于第一有机溶剂中后反应,分离得到化合物3;
s71.将化合物3与氨基苄氧羰基保护的左旋缬氨酸溶解于所述第二有机溶剂后,依次添加三乙胺和2,4,6-三氯苯甲酰氯反应,再添加4-二甲氨基吡啶反应,分离得到化合物12;
s81.将所述化合物12和所述第一钯催化剂溶解于所述第三有机溶剂后反应,得到氨基醇粗产物;然后将所述氨基醇粗产物与6-庚烯酸溶解于所述第四有机溶剂中,依次添加n,n-二异丙基乙胺、1-羟基-7-偶氮苯并三氮唑和2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯后反应,分离得到化合物14;
s91.将所述化合物14、铱催化剂、4-氯-3-硝基苯甲酸和碳酸铯溶解于四氢呋喃、水和乙酸烯丙酯的混合溶液中反应,分离得到化合物2;
s111.将三乙胺和三氟甲磺酸三甲基硅酯依次添加到所述化合物2的溶液中反应,分离得到化合物2’;然后将所述化合物2’溶解在所述第七有机溶剂后添加格拉布斯二代催化剂反应,分离得到化合物15b;
s121.将所述化合物15和第二钯催化剂溶解于所述第八有机溶剂后反应,分离得到dysoxylactama。
为使本发明上述实施细节和操作能清楚地被本领域技术人员理解,以及本发明实施例十七元大环脂肽天然化合物的合成方法的进步性能显著的体现,本发明实施例1~10制备了十七元大环脂肽天然化合物,dysoxylactama,合成路线图如附图1所示。
实施例1
制备化合物6的步骤包括:在-40℃~-80℃将sbuli(asolutioninhexanes,1.3m,1.9ml,2.48mmol,2.0eq.)缓慢滴加到氨基甲酸酯5(411mg,1.24mmol,1.0eq.)和tmeda(0.41ml,2.73mmol,2.2eq.)的无水乙醚(15ml,0.08m)溶液中。在-40℃~-80℃反应5~8个小时然后缓慢滴加hbpin(0.54ml,3.72mmol,3.0eq.)的乙醚(2ml)溶液中。在-40℃~-80℃反应一到三个小时后撤去冷阱,反应液升温至30℃~50℃继续反应10~15个小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入kh2po4水溶液(1maq.,5ml)淬灭反应,继续搅拌10~30分钟。反应液用乙醚(3x10ml)萃取,合并得到的有机相先后用水(15ml)和饱和食盐水洗涤洗涤(20ml),再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状有机硼试剂化合物6(324mg,83%)。[α]d24=-1.5(c0.5,chcl3);1hnmr(300mhz,cdcl3):δ3.42(dd,j=9.6,5.8hz,1h),3.30(dd,j=9.6,7.1hz,1h),1.92–1.74(m,1h),1.23(s,12h),0.91(d,j=6.7hz,3h),0.88(s,9h),0.88–0.79(m,1h),0.57(dd,j=15.6,8.9hz,1h),0.02(s,6h).13cnmr(75mhz,cdcl3):δ83.0,70.2,32.4,26.1,25.0,24.9,19.1,18.5,-5.2.hrms(esi,m/z)forc16h36bo3si+[m+h]+:calcd.315.2521;found315.2527。
实施例2
制备化合物7的步骤包括:在-40℃~-80℃将sbuli(asolutioninhexanes,1.3m,0.74ml,0.96mmol,3.0eq.)缓慢滴加到乙基氨基甲酸酯(166mg,0.96mmol,3.0eq.)和(+)-sparteine(0.29ml,1.28mmol,4.0eq.)的无水乙醚(10ml,0.1m)溶液中。在-40℃~-80℃反应3~5个小时然后缓慢滴加化合物6(100mg,0.32mmol,1.0eq.)的乙醚(5ml)溶液中。在-40℃~-80℃反应一个到三个小时后撤去冷阱,反应液升温至30~50℃继续反应10~15个小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入kh2po4水溶液(1maq.,5ml)淬灭反应,继续搅拌10分钟。反应液用乙醚(3x10ml)萃取,合并得到的有机相先后用水(15ml)和饱和食盐水洗涤洗涤(20ml),再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状有机硼酸酯化合物7(99.4mg,91%,>20:1dr)。[α]d24=+2.8(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ3.47(dd,j=9.7,5.3hz,1h),3.31(dd,j=9.8,7.2hz,1h),1.70–1.56(m,1h),1.32–1.26(m,1h),1.22(s,12h),1.20–1.15(m,1h),1.12–1.01(m,1h),0.93(d,j=7.1hz,3h),0.88(s,9h),0.85(d,j=6.7hz,3h),0.02(s,6h).13cnmr(101mhz,cdcl3)δ82.9,68.8,36.4,34.6,26.1,24.9,24.9,18.5,16.8,15.4,-5.2,-5.2.hrms(m/z):calculatedforc14h30nao3si+[m+na]+:297.1862,found297.1862。
实施例3
制备化合物8的步骤包括:将有机硼酸酯7(99.4mg,0.29mmol,1.0eq.)和碘氯甲烷(0.42ml,5.8mmol,20.0eq.)溶于极其干燥的thf(20ml,0.15m)中,-40℃~-80℃条件下缓慢滴加正丁基锂溶液(2.5minhexanes,0.58ml,1.45mmol,5.0eq.),(如果滴加速度过快溶液会变成黄色)。随后将反应体系升至室温并持续搅拌10~15小时。反应结束后,将反应液冷却到0℃~25℃并向体系中加入2nnaoh/30%wth2o2水溶液(6ml/3ml)淬灭反应,继续搅拌30~50分钟。反应液用乙醚(3x10ml)萃取,合并得到的有机相先后用水(15ml)和饱和食盐水洗涤洗涤(20ml),再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状醇化合物8(64.5mg,90%)。[α]d23=-7.8(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ3.51–3.41(m,2h),3.41–3.37(m,2h),1.78(brs,1h)1.77–1.67(m,2h),1.26–1.06(m,2h),0.88(s,9h),0.88–0.87(m,3h),0.84(d,j=6.6hz,3h),0.03(s,6h).13cnmr(101mhz,cdcl3)δ69.2,69.1,37.0,33.2,33.1,26.1,18.5,16.8,16.6,-5.2.hrms(esi,m/z)forc18h40bo3si+[m+h]+:calcd.343.2834;found343.2836。
实施例4
制备化合物9的步骤包括:将化合物8(160mg,0.65mmol,1.0eq.)溶解于二氯甲烷(20ml,0.03m)中,冷却至0℃~5℃后依次加入nahco3(164mg,1.95mmol,3.0eq.),tcca(181mg,0.78mmol,1.2eq.)和tempo(10mg,0.065mmol,0.1eq.)。反应体系在0℃~5℃条件下搅拌2~5分钟后,反应液通过硅藻土过滤然后用na2s2o3水溶液淬灭反应。反应液用dcm(3x100ml)萃取,合并得到的有机相用饱和的nahco3水溶液(5ml),再用无水硫酸钠固体干燥。过滤之后,将滤液经真空水泵减压浓缩所得粗产物经硅胶柱层析分离即可得到无色油状化合物9(157mg,99%)。[α]d24=+1.7(c1.0,chcl3)1hnmr(300mhz,cdcl3)δ9.62(d,j=1.9hz,1h),3.43(dd,j=5.9,2.6hz,2h),2.52–2.36(m,1h),1.79–1.61(m,1h),1.58–1.33(m,2h),1.07(d,j=7.0hz,3h),0.89(s,9h),0.91–0.83(m,3h),0.03(s,6h).13cnmr(75mhz,cdcl3)δ205.6,68.3,44.3,34.1,33.4,26.1,18.5,16.6,13.6,-5.3.hrms(esi,m/z)forc13h28o2sina+[m+na]+:calcd.267.1751;found267.1760。
实施例5
制备化合物3的步骤包括:称取t-buok(280mg,2.5mmol,2.0eq.)于烧瓶中在80~100℃下抽真空过夜,然后加入thf(60ml,0.02m.)将其溶解,冷却到-60℃~-80℃加入顺式2-丁烯(350mg,6.2mmol,5.0eq.)随后逐滴滴加n-buli(1.0ml,2.5mmol,2.5minhexanes,2.0eq.).伴随着反应液变成橘黄色。然后升温至-30℃~-45℃反应10~30分钟然后再降温到-60℃~-80℃。紧接着在手套箱中称取(+)-ipc2bome(1.2g,3.8mmol,3.0eq.)并溶解在thf(10ml.)加入,逐滴滴加同时反应液变成亮黑色。10~30分钟后逐滴滴加bf3·oet2(0.6ml,5.0mmol,4.0eq.),紧接着将醛9(302mg,1.3mmol,1.0eq.)溶解在thf(5ml)中逐滴加入。然后在-60℃~-80℃反应10~15个小时,经tlc监测确认化合物9转化完全后用naoh(3m,30ml)将反应淬灭再加入过氧化氢(30%,20ml)在50~70℃回流1~2h搅拌至澄清。用乙醚萃取(3x300ml)合并有机相之后用饱和nh4cl溶液(30ml)洗涤,无水硫酸钠固体干燥。过滤,减压蒸馏除去有机溶剂,经快速柱层析分离以87%的产率得到无色油状产品化合物3(329mg,1.1mmol)。[α]d25=-9.9(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ5.79(ddd,j=17.2,10.5,7.3hz,1h),5.23–4.92(m,2h),3.62–3.29(m,2h),3.27–3.12(m,1h),2.47–2.33(m,1h),1.77–1.60(m,2h),1.37–1.14(m,2h),1.03(d,j=6.8hz,3h),0.89(s,9h),0.91–0.85(m,3h),0.83(d,j=6.6hz,3h),0.04(s,6h).13cnmr(101mhz,cdcl3)δ142.1,114.8,79.5,69.6,40.6,34.6,33.3,33.1,26.1,18.5,16.5,16.3,13.9,-5.2.hrms(esi,m/z)forc17h36o2sina+[m+na]+:calcd.323.2377;found323.2382。
实施例6
制备化合物12的步骤包括:将化合物3(329mg,1.1mmol,1.0eq.)和氨基cbz保护的l-缬氨酸(552mg,2.2mmol,2.0eq.)溶解于甲苯(110ml,0.01m)在零度的条件下依次加入三乙胺(0.61ml,4.4mmol,4.0eq.)和tcbc(0.52ml,3.3mmol,3.0eq.)然后反应液升温至室温继续搅拌10~30分钟后在零度条件下加入dmap。然后将反应液升温至室温反应10~12小时后经tlc监测确认转化完全后用饱和nh4cl水溶液(15ml)将反应淬灭,然后用乙酸乙酯萃取(3x10ml)合并有机相之后用饱和食盐水溶液(20ml)洗涤,无水硫酸钠固体干燥。过滤,减压蒸馏除去有机溶剂,经快速柱层析分离以91%的产率得到无色油状产品12(554mg,1.0mmol)。[α]d26=-2.1(c0.5,chcl3);1hnmr(300mhz,cdcl3)δ7.41–7.27(m,5h),5.67(ddd,j=17.6,10.2,7.8hz,1h),5.25(d,j=9.4hz,1h),5.11(s,2h),5.08–4.97(m,2h),4.81(dd,j=7.3,4.7hz,1h),4.32(dd,j=9.3,4.2hz,1h),3.37(dd,j=6.5,1.9hz,2h),2.58–2.46(m,1h),2.28–2.15(m,1h),1.93–1.77(m,1h)1.66–1.54(m,1h),1.27–1.19(m,1h),1.14–1.06(m,1h),1.01(d,j=6.9hz,3h),0.96(d,j=6.7hz,3h),0.89(s,9h),0.88–0.83(m,6h),0.79(d,j=6.6hz,3h),0.04(s,6h).13cnmr(75mhz,cdcl3)δ172.1,156.4,140.3,136.5,128.7,128.3,128.3,115.4,82.5,69.4,67.1,59.4,39.5,33.8,33.2,32.1,31.2,26.1,19.7,18.5,17.2,16.6,15.8,15.7,-5.2.hrms(esi,m/z)forc30h52no5sina+[m+na]+:calcd.556.3429;found556.3429。
实施例7
制备化合物14的步骤包括:将化合物12(187mg,0.35mmol,1.0eq.)溶解于甲醇溶液中(10ml,0.04m),在室温氮气保护下加入二氯化钯(25mg,0.14mmol,0.4eq.),随后将反应体系中的氮气置换成氢气。在室温下继续搅拌3~5小时(通过lc-ms检测到所有的保护基被脱除,双键全部被还原),将反应通过硅藻土过滤,滤液经旋转蒸发仪减压浓缩得到黄色油状氨基醇化合物13。不用进一步纯化直接用于下一步反应。将粗产品氨基醇和6-庚烯酸(90mg,0.70mmol,2.0eq.)溶解于dcm/dmf(20ml/1ml,0.018m)混合溶液中然后在零度条件下依次加入dipea(0.62ml,3.5mmol,10.0eq.),hoat(95mg,0.70mmol,2.0eq.)和hatu(532mg,1.4mmol,4.0eq.)。反应液升温至室温继续反应8~10个小时经tlc监测确认转化完全后用饱和nh4cl水溶液(5ml)将反应淬灭,然后用dcm萃取(3x20ml)合并有机相之后用无水硫酸钠固体干燥。过滤,减压蒸馏除去有机溶剂,经快速柱层析分离以两步58%的产率得到无色油状产品14(81mg,0.20mmol)。[α]d26=-8.5(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ5.87(d,j=9.0hz,1h),5.79(ddt,j=16.9,10.2,6.7hz,1h),5.00(dq,j=17.1,1.7hz,1h),4.97–4.91(m,1h),4.78(dd,j=8.0,3.7hz,1h),4.51(dd,j=8.7,4.6hz,1h),3.45(d,j=5.7hz,2h),2.24(t,j=7.6hz,2h),2.24–2.12(m,1h),2.13–2.00(m,2h),1.85(ddt,j=14.3,7.5,3.5hz,1h),1.76–1.61(m,4h),1.52–1.35(m,2h),1.36–1.28(m,1h),1.24–1.19(m,1h),1.17–1.09(m,2h),1.00(d,j=6.8hz,3h),0.95–0.83(m,15h).13cnmr(101mhz,cdcl3)δ173.4,172.6,138.6,114.8,82.4,69.2,57.6,36.6,36.0,35.3,33.5,33.0,31.9,30.7,28.6,27.0,25.2,19.8,17.7,16.0,15.5,13.7,11.9。hrms(esi,m/z)forc23h43no4na+[m+na]+:calcd.420.3084;found420.3083。
实施例8
制备化合物2的步骤包括:氮气保护下,将醇化合物14(66mg,0.16mmol,1.0eq.),铱催化剂(86mg,0.08mmol,0.5eq.),4-cn-3-nitrobenzoicacid(34mg,0.16mmol,1.0eq.),cesiumcarbonate(65mg,0.2mmol,1.2eq.)加入到封管中,然后通过氮气置换加入四氢呋喃溶剂(5ml,0.03m)紧接着加入蒸馏水(10μl,2v%)和乙酸烯丙酯(0.17ml,1.6mmol,10.0eq.).将封管封严实避免空气的接触然后加热到80~100℃反应48~72小时。经tlc监测确认转化完全后反应体系降温至室温然后用二氯甲烷(20ml)稀释,固体物质通过硅藻土过滤,滤液减压蒸馏出去有机溶剂得到黄色油状物质。将其吸附在硅胶上通过快速柱层析分离以71%的产率得到相应的醇2。[α]d26=-2.5(c0.5,chcl3);1hnmr(500mhz,cdcl3)δ5.94–5.70(m,3h),5.14–5.05(m,2h),5.00(dd,j=17.2,1.8hz,1h),4.94(dd,j=10.2,1.8hz,1h),4.77(dd,j=8.2,3.6hz,1h),4.47(dd,j=8.6,4.6hz,1h),3.48(td,j=6.6,3.3hz,1h),2.29–2.20(m,4h),2.21–2.15(m,1h),2.10–2.03(m,2h),1.87–1.79(m,1h),1.76–1.69(m,1h),1.68–1.61(m,3h),1.61–1.55(m,1h),1.48–1.38(m,3h),1.31(tt,j=13.4,7.4hz,1h),1.20–1.10(m,1h),1.01(d,j=6.9hz,3h),0.96–0.87(m,9h),0.86–0.82(m,6h).13cnmr(126mhz,cdcl3)δ173.6,172.6,138.6,136.0,117.3,114.8,82.3,75.3,57.8,39.3,36.6,35.9,34.8,33.6,32.0,30.5,28.6,27.1,25.2,19.8,17.8,15.7,13.6,12.1,11.9.hrms(esi,m/z)forc26h47no4na+[m+na]+:calcd.460.3397;found460.3397。
实施例9
制备化合物15b的步骤包括:将化合物2(26mg,0.059mmol,1.0eq.)溶于二氯甲烷(20ml,0.003m)并在零度条件下依次滴加三乙胺(0.16ml,1.2mmol,20.0eq.)和tmsotf(0.11ml,0.59mmol,10.0eq.),然后反应液在室温条件下反应3~5个小时。经tlc监测确认转化完全后反应体系用饱和氯化铵溶液(2ml)淬灭反应。反应液恢复至室温后用dcm萃取(2×10ml)并合并有机相,有机相用无水硫酸钠干燥,过滤并减压蒸馏除去低沸点有机溶剂组分。所得粗产物用硅胶柱层析(乙酸乙酯/正己烷=1:4)以92%的产率制得无色油状化合物2′(28mg,0.05mmol)。[α]d26=+1.6(c0.25,chcl3);1hnmr(500mhz,cdcl3)δ5.91(d,j=8.8hz,1h),5.86–5.70(m,2h),5.14–4.90(m,4h),4.78(dd,j=6.6,4.8hz,1h),4.63(dd,j=8.8,4.1hz,1h),3.49(dt,j=7.3,4.9hz,1h),2.24(t,j=7.5hz,3h),2.21–2.11(m,2h),2.11–2.02(m,2h),1.85–1.75(m,1h),1.73–1.63(m,3h),1.56(qd,j=6.7,4.5hz,1h),1.47–1.39(m,2h),1.34(ddd,j=13.3,7.6,5.8hz,1h),1.19–1.14(m,1h),1.11(dd,j=8.8,5.3hz,2h),0.98(d,j=6.9hz,3h),0.93–0.85(m,9h),0.85–0.80(m,6h),0.09(s,9h).13cnmr(126mhz,cdcl3)δ172.7,172.4,138.6,136.0,116.7,114.8,82.8,77.0,57.2,39.0,36.8,36.1,35.4,35.0,33.6,32.4,31.4,28.7,26.8,25.3,19.7,17.5,16.2,14.2,13.9,0.7.hrms(esi,m/z)forc29h55no4sina+[m+na]+:calcd.532.3793;found532.3791。
将二烯化合物2′(10mg,0.02mmol,1.0eq.)称取在一个充分干燥的50ml圆底烧瓶中然后安装上回流管通过氮气进行三次置换。然后加入提前进行除气干燥的二氯甲烷(20ml,0.001m),紧接着加入grubbs二代催化剂(1.6mg,0.002mmol,0.1eq.)溶解在提前进行除气干燥的二氯甲烷(5ml)的溶液。然后将圆底烧瓶浸没在30~40℃的油浴锅中,并在此温度下反应20~25小时。经tlc监测确认转化完全后反应体系暴露在空气中搅拌10~30分钟然后通过旋转蒸发仪除去低沸点的有机溶剂得到深棕色的油状物质化合物15b,不用进一步纯化直接用于下一步反应。
实施例10
制备化合物dysoxylactama的步骤包括:将粗产品15b溶解于甲醇溶液中(10ml,0.002m),在室温氮气保护下加入二氯化钯(2mg,0.01mmol,0.5eq.),随后将反应体系中的氮气置换成氢气。在室温下继续搅拌3~4小时(通过lc-ms检测到tms保护基被脱除,双键全部被还原),将反应通过硅藻土过滤,滤液经旋转蒸发仪减压浓缩得到的粗产品吸附在柱层析硅胶上经硅胶柱层析(乙酸乙酯/正己烷=1:2)纯化以70%的产率通过两步得到白色固体天然产物dysoxylactama(5.8mg,0.014mmol)。[α]d22=-3.6(c1.0,chcl3);1hnmr(400mhz,pyridine-d5):δ5.69(brs,1h),5.18(dd,j=9.4,4.8hz,1h),5.08(dd,j=8.6,3.1hz,1h),3.69(ddd,j=7.8,5.3,2.3hz,1h),2.61–2.45(m,2h),2.40–2.28(m,1h),2.13–2.03(m,1h),2.03–1.96(m,1h),1.95–1.88(m,1h),1.86–1.79(m,2h),1.76–1.68(m,1h),1.67–1.61(m,2h),1.61–1.56(m,1h),1.56–1.52(m,1h),1.49–1.43(m,2h),1.43–1.40(m,1h),1.39–1.35(m,1h),1.35–1.28(m,1h),1.28–1.18(m,2h),1.15(d,j=6.7hz,3h),1.08(d,j=6.8hz,3h),1.00(d,j=6.9hz,3h),0.99(d,j=6.7hz,3h),0.95–0.89(m,6h).13cnmr(101mhz,pyridine-d5):δ173.7,81.6,76.4,57.9,37.8,36.4,36.0,35.1,33.2,33.0,31.8,28.7,28.2,28.1,27.7,25.9,25.3,20.2,18.1,16.8,16.3,13.9,12.3.hrms(esi,m/z)forc24h45no4na+[m+na]+:calcd.434.3241;found434.3224。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!