一种从金钗石斛中提取化合物的工艺及应用的制作方法
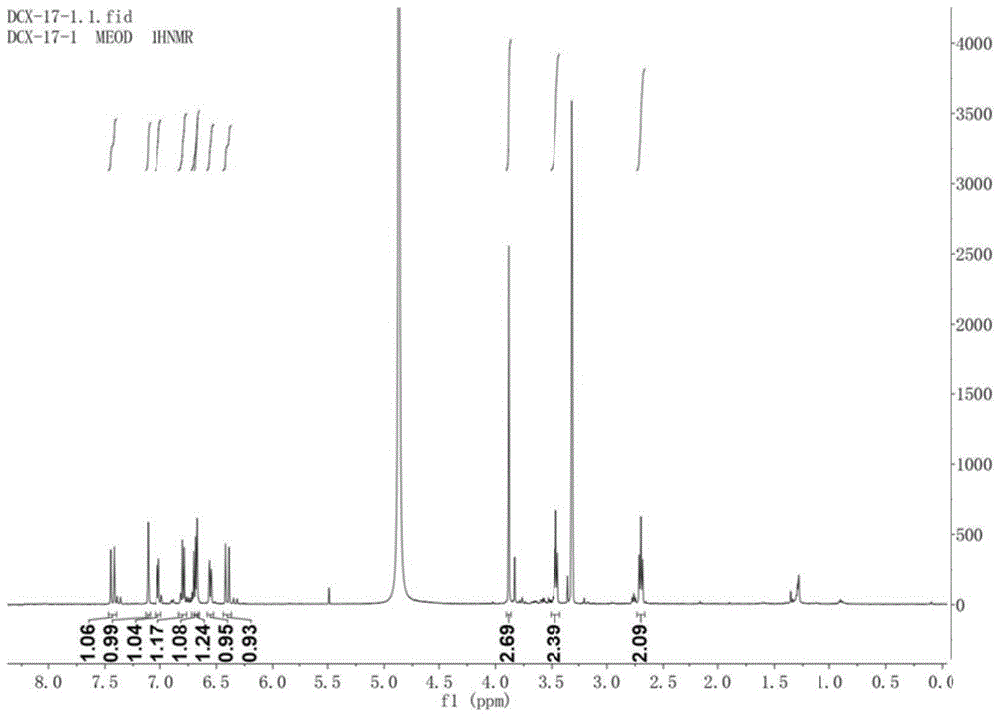
本发明属于生物
技术领域:
,尤其涉及一种从金钗石斛中提取化合物的工艺及应用。
背景技术:
:近年来,随着人们生活水平的提高,糖尿病的患病率也在逐年增加,据2017年国际糖尿病联盟(idf)发布的数据显示,全世界约有4.25亿人(20-79岁)患有糖尿病,其中中国有1.14亿,位居世界第一,根据这一趋势,到2045年,全球将有6.29亿人是糖尿病患者。此外,糖尿病的发病率高达12%,并且在逐渐趋于年轻化。糖尿病(diabetesmellitus,dm)是一种多种因素和病因造成的代谢紊乱疾病,它的特征在于慢性的高血糖症。其发病与饮食、遗传、环境因素和免疫系统功能紊乱等有密切关系。糖尿病主要分为三类,i型、ii型和妊娠糖尿病,其中ii型患者占总糖尿人数的90%以上,但目前对其发生发展的精确分子机制尚不完全清楚。近几年,积累的诸多临床实验证据揭示ii型糖尿病的发病机制与肥胖及炎症所引起的胰岛素抵抗有关。炎症是机体、细胞对外界物理、化学和生物等有害刺激做出的一系列免疫应答反应,反应的常见表现有发热、红肿、疼痛以及功能障碍等。一般来说,炎症是组织器官对损伤或者感染做出的防御反应,对机体是有益的,但是长期的炎症反应会诱导机体产生过激的异常反应,甚至会诱发许多疾病,如糖尿病、动脉粥样硬化、老年痴呆症、癌症等。目前临床上常使用甾体激素和非甾体类(阿司匹林、双氯芬酸和布洛芬等)治疗炎症,人体服用后会产生一系列的毒副作用,如肝脏和胃肠道损伤以及心血管系统损伤等。此外,植物源药物药效缓慢温和、持久性强,毒副作用小且不易产生机体耐受,因此有必要从植物中寻找具有降血糖和抗炎活性的物质,为新型糖尿病和炎症药物的研发提供先导化合物。技术实现要素:本发明的目的在于提供一种从金钗石斛中提取化合物的工艺及应用,本发明中的工艺从药用植物金钗石斛的茎中提取得到式i和ii化合物,能有效抑制血糖水平关键酶的活性,以lps诱导raw264.7产生no为模型进行抗炎活性评价,i和ii所示的化合物能够抑制no的产生,体现抗炎活性。本发明提供一种从金钗石斛中提取化合物的工艺,包括以下步骤:a)将金钗石斛粉碎,用1~3倍体积的乙醇水溶液浸提2~5次,所得浸提液过滤后合并浓缩,得到浸膏a;b)将所述浸膏a与水制备成混悬液,依次用石油醚、乙酸乙酯和正丁醇进行萃取,筛选三种萃取液后,将乙酸乙酯萃取液浓缩制成浸膏b;c)取浸膏b,过减压柱,经石油醚和乙酸乙酯混合洗液梯度洗脱,然后用丙酮冲柱,收集流分,将所得各个流分浓缩合并,最终获得16个流分,记为fr.1~fr.16;d)将流分fr.13过反相柱,以30~100%的甲醇水溶液进行梯度洗脱,得到14个流分,记为fr.13-1~fr.13-14;e)取流分fr.13-7过葡聚糖凝胶柱,以甲醇和氯仿的混合液进行洗脱,得到18个流分,记为fr.13-7-1~fr.13-7-18,合并fr.13-7-15~fr.13-7-17流分,经半制备hplc纯化,在tr=13.5min得到式i所示化合物,在tr=16.4min得到式ii所示化合物;优选的,所述步骤b)中浸膏a与水的体积比为1∶(0.5~2);所述浸膏a与石油醚的体积比为1∶(0.5~2);所述浸膏a与乙酸乙酯的体积比为1∶(0.5~2);所述浸膏a与正丁醇的体积比为1∶(0.5~2)。优选的,所述步骤c)中石油醚和乙酸乙酯混合洗液的梯度洗脱具体为:石油醚和乙酸乙酯的体积比在80~120小时内由20∶1均匀降至0∶1,所述梯度洗脱的总时间优选为90~110小时,更优选为100小时。优选的,所述步骤c)将得到的各个流分减压浓缩,经过薄层层析点板检测,根据显色合并相似流分。优选的,所述步骤d)中梯度洗脱具体为:依次以质量浓度30%、40%、50%、60%、70%、80%、90%和100%的甲醇水溶液进行洗脱,每个洗脱梯度的洗脱时间相等,洗脱总时间为24~48小时,更优选为32~36小时。优选的,所述步骤e)中甲醇与氯仿的体积比为1∶(0.5~2)。本发明提供一种如上文所述的工艺提取得到的式i所示化合物和式ii所示化合物在制备防治糖尿病和缓解炎症的药物中的应用。优选的,所述防治糖尿病的药物为α-葡萄糖苷酶活性抑制剂;所述缓解炎症的药物为no的产生抑制剂。本发明提供一种药物制剂,包括上文所述的工艺提取得到的式i所示化合物或式ii所示化合物以及药学上可接受的辅料。优选的,所述药物制剂的剂型为片剂、胶囊剂、丸剂、颗粒剂、汤剂、膏剂、露剂、口服液剂、滴丸剂或糖浆剂。本发明提供了一种从金钗石斛中提取化合物的工艺,包括以下步骤:a)将金钗石斛粉碎,用1~3倍体积的乙醇水溶液浸提2~5次,所得浸提液过滤后合并浓缩,得到浸膏a;b)将所述浸膏a与水制备成混悬液,依次用石油醚、乙酸乙酯和正丁醇进行萃取,将乙酸乙酯萃取液浓缩制成浸膏b;c)取浸膏b,过减压柱,经石油醚和乙酸乙酯混合洗液梯度洗脱,最后用丙酮冲柱,收集流分,将所得各个流分浓缩合并,最终获得16个流分,记为fr.1~fr.16;d)将流分fr.13过反相柱,以30~100%的甲醇水溶液进行梯度洗脱,得到14个流分,记为fr.13-1~fr.13-14;e)取fr.13-7过葡聚糖凝胶柱,以甲醇和氯仿的混合液进行洗脱,得到18个流分,记为fr.13-7-1~fr.13-7-18,合并fr.13-7-15~fr.13-7-17流分,经半制备hplc纯化,在tr=13.5min得到式i所示化合物,在tr=16.4min得到式ii所示化合物。本发明研究表明式i和ii所示的化合物能够抑制α-葡萄糖苷酶的活性,以lps诱导raw264.7产生no为模型进行抗炎活性评价,i和ii所示的化合物能够抑制no的产生,体现抗炎活性。因此表明该化合物能够用于制备治疗和/或预防糖尿病和炎症的食品和/或药品。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。图1是本发明实施例1中式i所示化合物的1hnmr图谱;图2是本发明实施例1中式i所示化合物的13cnmr+dept135图谱;图3是本发明实施例1中式i所示化合物的hsqc图谱;图4是本发明实施例1中式ii所示化合物的1hnmr图谱;图5是本发明实施例1中式ii所示化合物的13cnmr+dept135图谱;图6是本发明实施例1中式ii所示化合物的hsqc图谱。具体实施方式本发明提供了一种从金钗石斛中提取化合物的工艺,包括以下步骤:a)将金钗石斛粉碎,用1~3倍体积的乙醇水溶液浸提2~5次,所得浸提液过滤后合并浓缩,得到浸膏a;b)将所述浸膏a与水制备成混悬液,依次用石油醚、乙酸乙酯和正丁醇进行萃取,筛选三种萃取液后,将乙酸乙酯萃取液浓缩制成浸膏b;c)取浸膏b,过减压柱,经石油醚和乙酸乙酯混合洗液梯度洗脱,最后用丙酮冲柱,收集流分,将所得各个流分浓缩合并,最终获得16个流分,记为fr.1~fr.16;d)将流分fr.13过反相柱,以30~100%的甲醇水溶液进行梯度洗脱,得到14个流分,记为fr.13-1~fr.13-14;e)取fr.13-7过葡聚糖凝胶柱,以甲醇和氯仿的混合液进行洗脱,得到18个流分,记为fr.13-7-1~fr.13-7-18,合并fr.13-7-15~fr.13-7-17流分,经半制备hplc纯化,在tr=13.5min得到式i所示化合物,在tr=16.4min得到式ii所示化合物;金钗石斛(dendrobiumnobilelindl.)为兰科(orchidaceae)石斛属(dendrobiumsw.)植物,又名金钗石、扁金钗、扁黄草等,为我国传统名贵中药,素有“千金草”之称,常以新鲜或干燥茎入药。金钗石斛在我国主要分布于贵州、云南、广西等长江以南的亚热带地区。在本发明中,优选使用金钗石斛的干燥茎为原料进行提取。本发明将金钗石斛干燥茎粉碎,加乙醇水溶液浸提,所得浸提液过滤后合并浓缩成浸膏a。在本发明中,所述金钗石斛干燥茎的粉碎粒径优选为0.1~1cm,更优选为0.5~0.6cm;所述乙醇水溶液中乙醇与水的体积比优选为(15~20)∶1,更优选为(18~19)∶1;所述乙醇水溶液的体积为粉碎后的金钗石斛的体积的1~3倍,优选为2倍体积;所述浸提的次数优选为2~5次,优选为3~4次;所述浸提液浓缩的浓缩比优选为(3~5)∶1,更优选为4∶1。得到浸膏a后,本发明将所述浸膏a与水制备成混悬液,然后依次用石油醚、乙酸乙酯和正丁醇进行萃取,各萃取层萃取至萃取液无色停止萃取,将乙酸乙酯萃取液浓缩制成浸膏b。石油醚、乙酸乙酯和正丁醇极性依次增大,正丁醇萃取液中可能是一些极性大的糖苷类化合物,具体实验时选择极性中等的流分,即乙酸乙酯萃取液。在本发明中,所述浸膏a与水的体积比优选为1∶(0.5~2),更优选为1∶(1~1.5);所述浸膏a与石油醚的体积比为1∶(0.5~2),更优选为1∶(1~1.5);具体的,在本发明的实施例中,浸膏a∶水∶石油醚的体积比=1∶1∶1;所述浸膏a与乙酸乙酯的体积比优选为1∶(0.5~2),更优选为1∶(1~1.5);具体的,在本发明的实施例中,浸膏a∶水∶乙酸乙酯的体积比=1∶1∶1;所述浸膏a与正丁醇的体积比为1∶(0.5~2),更优选为1∶(1~1.5)。具体的,本发明的实施例中,浸膏a∶水∶正丁醇的体积比=1∶1∶1。在本发明中,所述乙酸乙酯萃取液的浓缩比优选为(2~5)∶1,更优选为(3~4)∶1。得到浸膏b后,本发明将浸膏b过减压柱,经石油醚和乙酸乙酯混合洗液梯度洗脱,然后用丙酮冲柱,收集流分,将所得各个流分浓缩合并,最终获得16个流分,记为fr.1~fr.16。在本发明中,所述减压柱为硅胶柱层析,所述硅胶柱为硅胶h,所述硅胶h的粒径为100~200目。所述石油醚与乙酸乙酯的混合溶液的体积比为(20~0)∶1,具体的,在本发明的实施例中,所述梯度洗脱的具体过程为:石油醚与乙酸乙酯的混合溶液的体积比初始为20∶1,随着梯度洗脱的进行体积比均匀减小,直至降为0∶1,所述梯度洗脱的总时间优选为80~120小时,更优选为90~110小时,最优选为100小时;所述梯度洗脱的温度优选为室温,即20~35℃,优选为25~30℃,每个梯度所用洗脱液的体积优选为3~4l。洗脱完成后,使用丙酮冲柱,收集流分,优选的,每500ml收集一次,将所得各个流分经过薄层层析(tlc)点板检测,合并相似流分,获得16个流分,记为fr.1~fr.16。在本发明中,得到的各个流分经过薄层层析(tlc)点板检测,肉眼观察,主点相同或者类似的合并到一起,共获得16个流分。经高效液相色谱分析后,fr.13特征峰比较明显,将其过反相柱(填料为c18),依次以30%、40%、50%、60%、70%、80%、90%、100%甲醇水溶液梯度洗脱,100ml收集一次,每个梯度用1.5l混合液,所得各个流分经过薄层层析(tlc)点板检测,肉眼观察,主点相同或者类似的合并到一起,得到14个流份,记为fr.13-1~fr.13-14。在本发明中,所述梯度洗脱中,每个梯度的洗脱时间相等,所述洗脱的总时间优选为24~48小时,更优选为32~36小时。取流分fr.13-7过葡聚糖sephadexlh-20凝胶柱,以甲醇和氯仿的混合液进行洗脱,室温条件下,2~3秒一滴,每5ml收集一管,所得各个流分减压浓缩,经过薄层层析(tlc)点板检测,合并相似流分,得到18个流分,记为fr.13-7-1~fr.13-7-18。在本发明中,所述甲醇和氯仿的体积比优选为1∶(0.5~2),更优选为1∶(1~1.5)。fr.13-7-15~fr.13-7-17呈点相似,将其合并后经半制备hplc(c18色谱柱,35%甲醇-水洗脱)纯化,得到式i所示化合物(tr=13.5min)和式ii化合物(tr=16.4min)。本发明还提供了一种式i所示化合物或式ii所示化合物在制备防治糖尿病和缓解炎症的药物中的应用,优选的,所述防治糖尿病的药物为α-葡萄糖苷酶活性抑制剂,所述糖尿病为ii型糖尿病,本发明中的式i和式ii所示化合物通过抑制α-葡萄糖苷酶的活性,减少低聚糖在消化道内分解,延缓肠道对葡萄糖的吸收,从而降低餐后高血糖风险,实现降低血糖水平的效果;本发明中所述缓解炎症的药物为no的产生抑制剂,本发明中的式i所示化合物或式ii所示化合物能够抑制no的产生,体现抗炎活性,缓解机体发红、肿胀、发热、疼痛等症状。本发明还提供了一种药物制剂,包括上述工艺提取得到的式i所示化合物或式ii所示化合物以及药学上可接受的辅料。优选的,所述药物制剂的剂型优选为口服制剂,更优选为片剂、胶囊剂、丸剂、颗粒剂、汤剂、膏剂、露剂、口服液剂、滴丸剂或糖浆剂。更优选的,所述胶囊剂为硬胶囊剂或软胶囊剂;更优选的,片剂为口服片剂或口腔片剂;更优选的,口服片剂指供口服的片剂,多数此类片剂中的药物是经胃肠道吸收而发挥作用,也有的片剂中的药物是在胃肠道局部发挥作用。在本发明提供的一些实施例中,口服片剂为普通压制片、分散片、泡腾片、咀嚼片、包衣片或缓控释片。所述药学上可接受的辅料包括水果粉、食用香精、甜味剂、酸味剂、填充剂、润滑剂、防腐剂、助悬剂、食用色素、稀释剂、乳化剂、崩解剂或增塑剂中的一种或两者以上的混合物。本发明提供了一种从金钗石斛中提取化合物的工艺,包括以下步骤:a)将金钗石斛粉碎,用1~3倍体积的乙醇水溶液浸提2~5次,所得浸提液过滤后合并浓缩,得到浸膏a;b)将所述浸膏a与水制备成混悬液,依次用石油醚、乙酸乙酯和正丁醇进行萃取,将乙酸乙酯萃取液浓缩制成浸膏b;c)取浸膏b,过减压柱,经石油醚和乙酸乙酯混合洗液梯度洗脱,最后用丙酮冲柱,收集流分,将所得各个流分浓缩合并,最终获得16个流分,记为fr.1~fr.16;d)将流分fr.13过反相柱,以30~100%的甲醇水溶液进行梯度洗脱,得到14个流分,记为fr.13-1~fr.13-14;e)取fr.13-7过葡聚糖凝胶柱,以甲醇和氯仿的混合液进行洗脱,得到18个流分,记为fr.13-7-1~fr.13-7-18,合并fr.13-7-15~fr.13-7-17流分,经半制备hplc纯化,在tr=13.5min得到式i所示化合物,在tr=16.4min得到式ii所示化合物。本发明研究表明式i和ii所示的化合物能够抑制α-葡萄糖苷酶的活性,以lps诱导raw264.7产生no为模型进行抗炎活性评价,i和ii所示的化合物能够抑制no的产生,体现抗炎活性。因此表明该化合物能够用于制备治疗和/或预防糖尿病和炎症的食品和/或药品。为了进一步说明本发明,以下结合实施例对本发明提供的一种从金钗石斛中提取化合物的工艺及应用进行详细描述,但不能将其理解为对本发明保护范围的限定。实施例1步骤1:将金钗石斛干燥茎(13kg)粉碎后,用2倍体积的乙醇水溶液浸提3次,所得浸提液过滤后合并浓缩成浸膏a;步骤2:将所述浸膏a与水体积比1∶1制成混悬液,依次用石油醚(浸膏a∶水∶石油醚=1∶1∶1)、乙酸乙酯(浸膏a∶水∶乙酸乙酯=1∶1∶1)、正丁醇(浸膏a∶水∶正丁醇=1∶1∶1)进行萃取,各萃取层萃取至萃取液无色停止萃取,将乙酸乙酯萃取液浓缩制成浸膏b;步骤3:取浸膏b,过减压柱,经石油醚、乙酸乙酯(20∶1→0∶1,v/v)的混合液梯度洗脱,最后用丙酮冲柱,每500ml收集一瓶,所得各个流分经过薄层层析(tlc)点板检测,合并相似流分,获得16个流分,记为fr.1~fr.16;步骤4:经高效液相色谱分析后,fr.13特征峰比较明显,将其过反相柱,以30%、40%、50%、60%、70%、80%、90%、100%甲醇水溶液梯度洗脱,100ml收集一瓶,每个梯度用1.5l混合液,所得各个流分经过薄层层析(tlc)点板检测,合并相似流分,得到14个流份,记为fr.13-1~fr.13-14;步骤5:fr.13-7呈点性较好,取其过葡聚糖凝胶柱,以500ml甲醇∶氯仿=1∶1的混合液进行洗脱,6ml收集一管,点板合并相似流分,得到18个流份,记为fr.13-7-1~fr.13-7-18;步骤6:fr.13-7-15~fr.13-7-17呈点相似,合并后经半制备hplc(c18色谱柱,35%甲醇-水洗脱)纯化,得到式i和ii化合物(tr=13.5min和tr=16.4min)。式i和式ii所示结构化合物的鉴定图谱如图1~6所示。式i和ii所示结构鉴定数据如下:低分辨质谱m/z:352.4354[m+na]+,分子式为c18h19no5;1hnmr(500mhz)和13cnmr(125mhz)数据如表1所示:表1式i和ii所示化合物的nmr数据(溶剂为氘代甲醇)式i和ⅱ化合物对α-葡萄糖苷酶的抑制作用待测样品溶液的配制:取450μl配置好的2u/ml的α-葡萄糖苷酶溶液(用ph=6.8的pbs溶液稀释)于ep管中,将待测化合物溶解于dmso后(浓度为2mm),取45μl的该溶液加至ep管中,摇匀,分别取4次110μl混合均匀的待测溶液于96孔板中;(阴性与空白:取450μl配置好的2u/ml的α-葡萄糖苷酶溶液于ep管中,再取45μl的dmso溶液加至ep管中,摇匀,分别取4次110μl混合均匀的待测溶液于96孔板中)将96孔板于37℃放置15分钟后,各组均加入40μl(2.5mmol/l的4-硝基苯基-β-d-吡喃葡萄糖苷)pnpg溶液;(空白:加入40μl(0.1mol/l)pbs溶液)将96孔板于37℃放置15分钟后,设置酶标仪于405nm波长下测量每孔的od值吸光度.计算化合物对α-糖苷酶的抑制活性,计算公式如下:抑制率=(oddmso-od样)/(oddmso-odpbs)×100%将化合物倍半稀释几个梯度后,以相同的检测方法检测不同浓度样品对α-葡萄糖苷酶的抑制率,利用graphpadprism7软件计算其ic50值。结果如表2所示,化合物对α-葡萄糖苷酶有一定程度的抑制活性且好于阳性对照阿卡波糖。表2化合物对α-葡萄糖苷酶的抑制活性测试样品ic50±sd(μm)金雀异黄酮(阳性对照)8.54±0.69阿卡波糖(阳性对照)701.45±0.52化合物i22.98±1.46化合物ii46.42±1.12式i和ⅱ化合物抗炎活性评价选取raw264.7(小鼠单核巨噬细胞白血病细胞),在96孔平底细胞培养板上接种100μl浓度为5×104个/ml的细胞,培养于37℃,5%co2,90%以上湿度的条件下,24h后加入50μl配制好的待测化合物溶液,继续在该条件下培养,1h后加入50μl配制的lps(终浓度500ng/ml)溶液,24h后每孔取上清100μl于新的96孔板中,之后向每孔加入100μl(40mg/ml)的griess试剂,十字交叉法混匀。于酶标仪540nm波长下测定并记录每孔的吸光度,按下面公式计算no抑制率。对照组为吲哚美辛,阴性对照组为dmso,把待测化合物倍半稀释5个浓度梯度。用横坐标表示待测化合物浓度,纵坐标表示抑制率,作图求出待测化合物的ic50值。抑制率(%)=(c2-c1)/(c2-c0)×100%;式中:c0、c1、c2分别为540nm下测得的空白对照组(不加lps)、实验组、阴性(加lps)对照组的吸光值。计算各浓度下的抑制率并绘制化合物浓度-抑制率曲线图,计算得到化合物对lps诱导raw264.7产生no的半抑制浓度(ic50值)。结果如表3所示,化合物能有效抑制raw264.7细胞no的产生量,表现出一定的抗炎活性且效果显著好于阳性对照吲哚美辛。表3化合物对raw264.7产生no的抑制作用测试样品ic50±sd(μm)吲哚美辛(阳性对照)52.30±2.11化合物i11.07±0.61化合物ii10.43±1.17以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1