一种特异性扩增RNA的引物、其设计方法及用途与流程
一种特异性扩增rna的引物、其设计方法及用途
技术领域
1.本发明涉及分子生物学领域。具体而言,本发明涉及一种反转录引物及其设计方法。通过在rt-pcr中使用这种引物,可以特异性扩增rna,而不会扩增相同序列的dna。
背景技术:
2.在转录过程中,以dna的一条链为模板,以碱基互补配对为原则,形成一条rna单链,其主要功能是实现遗传信息在蛋白质层面的表达,是遗传信息向表型转化过程的重要桥梁,所以rna检测对于基因表达与功能分析具有十分重要的意义。但是,由于rna的形成机制,转录得到的rna的序列必然会与dna的某一条链完全相同(原核生物)或部分完全相同(真核生物)。因此在dna和rna同时存在的核酸样品中,以反转录-聚合酶链式反应(rt-pcr)扩增为基础的rna检测手段可以同时扩增相同序列的dna模板,最终的扩增后产物既可能来自于rna模板,也可能来自于dna模板,这样难以对样品中的rna进行准确的定性或定量检测,该现象在检测原核生物rna或不跨越内含子的真核生物rna时会难以避免。
3.为消除dna模板对rna检测的干扰,在常规检测中多是采用脱氧核糖核酸酶(dnase i)对核酸样品进行消化以去除dna,并辅以琼脂糖凝胶电泳进行dna条带检测(原核生物),或采用跨内含子-外显子引物对核酸酶消化后的模板进行pcr扩增(真核生物),以确保无dna干扰。由于dna消化过程需要一定的时间(10-20分钟)和温度(37℃),即便是在无核糖核酸酶存在的溶液中,dna消化过程也很难保证rna的完整性,这也会在一定程度上影响rna检测的准确性。因此,一种不需要进行dna消化步骤,可以选择性扩增rna模板的一步法反转录pcr对于rna和dna共存环境下的rna检测具有十分重要的意义。
技术实现要素:
4.本发明设计并提供了一种反转录引物,在该引物的5’端人为引入一定数量的错配碱基。在反转录过程中利用该引物以rna为模板进行cdna第一链的合成。由于反转录温度较低,5’端的错配不会影响反转录引物对模板的识别与延伸,使得cdna的5’端出现与dna完全不同的碱基序列,在进入第一轮pcr扩增时,由正向引物引导合成cdna互补链,形成包含人为引入的错配部分的双链dna。当这种dna与基因组dna共同存在于pcr环境中时,反转录引物在一定的退火温度条件下只能退火结合与自身完全匹配的模板链,即包含人为引入的错配部分的dna模板,而不能与原始基因组dna模板进行正常的退火结合。对于原始基因组dna模板,即便存在与自身dna模板完全匹配的上游引物,也只能发生线性扩增。如此也就实现了通过引物设计在一步法反转录pcr中区分dna和rna模板的目的,在保证rna检测结果准确性的前提下,去除了dna消化过程,降低了检测试剂成本,加快了实验流程(如图1所示)。
5.第一方面,本发明提供了一种以rna为模板的反转录引物,其由rna模板互补部分和人为引入的错配部分组成。
6.第二方面,本发明提供了一种设计并获得第一方面的反转录引物的方法,包括以下步骤:设计引物,使其由rna模板互补部分和人为引入的错配部分组成。
7.第三方面,本发明提供了第一方面的反转录引物用于区分dna模板和rna模板,实现rna特异性扩增的用途,所述dna模板和rna模板存在于同一样品中或不同样品中。
8.第四方面,本发明提供了一种在同时存在dna和rna的样品中特异性扩增rna的方法,包括使用第一方面的反转录引物进行rt-pcr。
9.第五方面,本发明提供了一种rt-pcr试剂盒,其包含第一方面的反转录引物。
附图说明
10.图1为使用本发明的反转录引物,进行rt-pcr的原理示意图。
11.图2至图17为本发明的反转录引物的具体实施方案。
12.图18为以hpv 16型e6/e7 rna为模板的一步法rt-pcr产物的毛细管电泳结果。
13.图19为以hpv 16型e6/e7 dna为模板的一步法rt-pcr产物的毛细管电泳结果。
14.图20为以hpv 18型e6/e7 rna为模板的一步法rt-pcr产物的毛细管电泳结果。
15.图21为以hpv 18型e6/e7 dna为模板的一步法rt-pcr产物的毛细管电泳结果。
16.图22为以rna样本3-1为模板的一步法rt-pcr产物的毛细管电泳结果。
17.图23为以rna样本3-1为模板的常规pcr产物的毛细管电泳结果。
18.图24为以rna样本3-2为模板的一步法rt-pcr产物的毛细管电泳结果。
19.图25为以rna样本3-2为模板的常规pcr产物的毛细管电泳结果。
20.图26为以rna样本3-3为模板的一步法rt-pcr产物的毛细管电泳结果。
21.图27为以rna样本3-3为模板的pcr常规产物的毛细管电泳结果。
22.图28为以rna样本3-4为模板的一步法rt-pcr产物的毛细管电泳结果。
23.图29为以rna样本3-4为模板的常规pcr产物的毛细管电泳结果。
24.图30为以dna样本3-1为模板的一步法rt-pcr产物的毛细管电泳结果。
25.图31为以dna样本3-1为模板的常规pcr产物的毛细管电泳结果。
26.图32为以dna样本3-2为模板的一步法rt-pcr产物的毛细管电泳结果。
27.图33为以dna样本3-2为模板的常规pcr产物的毛细管电泳结果。
28.图34为以rna样本4-1为模板的一步法rt-pcr产物的毛细管电泳结果。
29.图35为以rna样本4-1为模板的常规pcr产物的毛细管电泳结果。
30.图36为以rna样本4-2为模板的一步法rt-pcr产物的毛细管电泳结果。
31.图37为以rna样本4-2为模板的常规pcr产物的毛细管电泳结果。
32.图38为以rna样本4-3为模板的一步法rt-pcr产物的毛细管电泳结果。
33.图39为以rna样本4-3为模板的常规pcr产物的毛细管电泳结果。
34.图40为以rna样本4-4为模板的一步法rt-pcr产物的毛细管电泳结果。
35.图41为以rna样本4-4为模板的常规pcr产物的毛细管电泳结果。
36.图42为以dna样本4-1为模板的一步法rt-pcr产物的毛细管电泳结果。
37.图43为以dna样本4-1为模板的常规pcr产物的毛细管电泳结果。
38.图44为以dna样本4-2为模板的一步法rt-pcr产物的毛细管电泳结果。
39.图45为以dna样本4-2为模板的常规pcr产物的毛细管电泳结果。
40.图46为以rna样本5-1为模板的rt-qpcr扩增曲线(fam荧光通道)
41.图47为以rna样本5-1为模板的rt-qpcr扩增曲线(vic荧光通道)
42.图48为以dna样本5-1为模板的rt-qpcr扩增曲线(fam荧光通道)
43.图49为以dna样本5-1为模板的rt-qpcr扩增曲线(vic荧光通道)
具体实施方式
44.本发明提供了一种以rna为模板的反转录引物,其由rna模板互补部分和人为引入的错配部分组成;
45.其中,所述反转录引物长度优选为20-70bp,更优选为20-60bp,最优选为25-50bp;gc含量优选为20%-80%,更优选为25%-65%,最优选为30%-60%;tm值为65℃-70℃,不存在任何发夹结构;
46.所述模板互补部分长度优选为6bp-25bp,更优选为10-20bp,最优选为13-16bp;gc含量优选为20%-80%,更优选为30%-70%,最优选为35%-65%;tm值为25℃-45℃,不存在任何发夹结构;
47.所述错配部分长度优选为5bp-64bp,更优选为7-30bp,最优选为10-15bp;gc含量优选为20%-80%,更优选为30%-70%,最优选为40%-60%;tm值为30℃-70℃,不存在任何发夹结构。
48.在一个实施方案中,所述rna模板互补部分与rna模板完全互补,不存在任何错配碱基;在其他实施方案中,所述rna模板互补部分与rna模板非完全互补,可存在错配碱基,优选地存在1-5个错配碱基,更优选1-3个错配碱基,最优选1个错配碱基,且该错配碱基不存在于所述引物3’端的5个碱基中。
49.在一个实施方案中,所述错配部分与rna模板完全错配,在对应碱基位置上不存在任何单独或连续的互补碱基;在其他实施方案中,所述错配部分与rna模板不完全错配,在对应碱基位置上可存在单独或连续的互补碱基,但互补碱基总数不超过错配部分碱基总数的40%。
50.优选地,所述引物序列为seq id no:2或seq id no:4。
51.在一个实施方案中,所述反转录引物还包含长度为3bp-20bp的连接用碱基;特别地,所述连接用碱基可以具有其他特殊用途。
52.优选地,所述引物序列为seq id no:6或seq id no:10。
53.在一个实施方案中,所述反转录引物还包含分别位于所述错配部分两侧的保护碱基部分和保护碱基互补部分,所述保护碱基部分长度为4bp-8bp且与rna模板完全错配,保护碱基部分与保护碱基互补部分为反向互补结构,其在50℃-55℃的退火温度下能够退火结合。
54.优选地,所述引物序列为seq id no:14。
55.另一方面,本发明提供了设计并获得所述反转录引物的方法,包括以下设计步骤:设计引物,使其由rna模板互补部分和人为引入的错配部分组成;
56.其中,所述反转录引物长度优选为20-70bp,更优选为20-60bp,最优选为25-50bp;gc含量优选为20%-80%,更优选为25%-65%,最优选为30%-60%;tm值为65℃-70℃,不存在任何发夹结构;
57.所述模板互补部分长度优选为6bp-25bp,更优选为10-20bp,最优选为13-16bp;gc含量优选为20%-80%,更优选为30%-70%,最优选为35%-65%;tm值为25℃-45℃,不存
在任何发夹结构;
58.所述错配部分长度优选为5bp-64bp,更优选为7-30bp,最优选为10-15bp;gc含量优选为20%-80%,更优选为30%-70%,最优选为40%-60%;tm值为30℃-70℃,不存在任何发夹结构。
59.在一个实施方案中,所述rna模板互补部分与rna模板完全互补,不存在任何错配碱基;在其他实施方案中,所述rna模板互补部分与rna模板非完全互补,可存在错配碱基,优选地存在1-5个错配碱基,更优选1-3个错配碱基,最优选1个错配碱基,且该错配碱基不存在于所述引物3’端的5个碱基中。
60.在一个实施方案中,所述错配部分与rna模板完全错配,在对应碱基位置上不存在任何单独或连续的互补碱基;在其他实施方案中,所述错配部分与rna模板不完全错配,在对应碱基位置上可存在单独或连续的互补碱基,但互补碱基总数不超过错配部分碱基总数的40%。优选地,所述引物序列为seq id no:2或seq id no:4。
61.在一个实施方案中,所述反转录引物还包含长度为3bp-20bp的连接用碱基;特别地,所述连接用碱基可以具有其他特殊用途;优选地,所述引物序列为seq id no:6或seq id no:10。
62.在一个实施方案中,所述反转录引物还包含分别位于所述错配部分两侧的保护碱基部分和保护碱基互补部分,所述保护碱基部分长度为4bp-8bp且与rna模板完全错配,保护碱基部分与保护碱基互补部分为反向互补结构,其在50℃-55℃的退火温度下能够退火结合;优选地,所述引物序列为seq id no:14。
63.另一方面,本发明还提供了所述反转录引物用于区分dna模板和rna模板,实现rna特异性扩增的用途,所述dna模板和rna模板存在于同一样品中或不同样品中。
64.另一方面,本发明还提供了一种在同时存在dna和rna的样品中特异性扩增rna的方法,包括使用所述反转录引物进行rt-pcr。
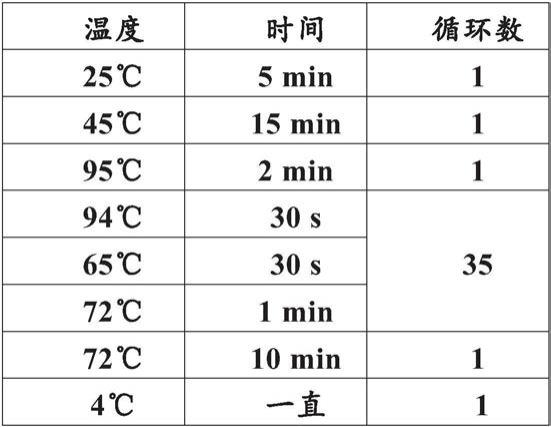
65.以下实施例用于示例性地说明本发明,而不对本发明进行限制。
66.实施例1:利用设计的引物进行一步法rt-pcr,区分人乳头瘤病毒(hpv)16型dna和rna模板
67.人乳头瘤病毒(hpv)高危型16型的持续感染是导致宫颈癌的重要原因。hpv 16型的e6/e7基因会被整合至人基因组中,导致e6/e7蛋白的大量表达,促进细胞转化,最终诱导癌症的发生。所以通过对e6/e7基因mrna的检测就能够更好地评估患者感染hpv的严重程度。
68.本实施例从宫颈脱落细胞中分别提取dna和rna,并在相同条件下分别进行一步法rt-pcr,最终利用毛细管电泳分离样品,检验本发明的引物对dna和rna模板的区分能力,具体步骤如下:
69.(1)根据图4中的方案设计hpv 16型e6/e7基因的反转录(反向)引物,并根据常规引物设计方案设计正向引物,并交由上海生工生物有限公司进行引物合成,其中正向引物5’端标记fam基团。具体的正向引物序列为5
’-
tgcccagctgtaatcatgcatggagatac-3’(seq id no:1),反转录(反向)引物序列为5
’-
cacactagagacatggcacaaccgaagc-3’(seq id no:2),其中小写字母部分为人为引入的错配碱基,大写字母部分为与rna模板互补部分,目标产物大小为234bp。
70.(2)获取确诊为hpv 16型感染患者的宫颈脱落细胞样本,并通过dna提取试剂盒和rna提取试剂盒分别对样本进行dna和rna提取,具体操作方法详见市售基因组dna提取试剂盒与rna提取试剂盒操作说明。需要注意最终用于一步法rt-pcr的dna样本需经过rnase a处理以去除rna残留,rna样本需经过dnase i处理以去除dna残留,并对dna和rna模板样本进行精确定量。
71.(3)在pcr八联管中分别加入dna和rna模板各2μl(浓度为1e+3拷贝/μl),rt-pcr酶0.5μl,上述正反向引物(1μm)各0.5μl,2
×
pcr缓冲液5μl,dutp(100mm)0.01μl,ddh2o补足反应体系10μl。
72.(4)将配制好的体系离心混匀,按表1程序进行rt-pcr反应。
73.表1 一步法rt-pcr反应程序
[0074][0075]
(5)准备电泳样品,每个电泳孔分别加入hi-di 8.7μl,size-5000.3μl,pcr产物1μl,混匀后离心备用。
[0076]
(6)毛细管电泳分离样品,将样品板置于3500dx基因分析仪中,选择“fragment”电泳方法,进行电泳,具体操作方法详见abi3500操作说明书。
[0077]
(7)结果分析。如图18所示,以提取的rna为模板的一步法rt-pcr毛细管电泳结果显示,一步法rt-pcr扩增出了片段长度为234bp的pcr产物,与设计引物时的目的片段大小一致。而图19则表明,以相同浓度dna为模板的一步法rt-pcr毛细管电泳结果显示,一步法rt-pcr并没有扩增出234bp的产物。由此可知,通过本发明设计的引物能够在一步法rt-pcr过程中实现对rna和dna的选择性扩增,能够很好的区分rna和dna模板。
[0078]
实施例2:利用设计的引物进行一步法rt-pcr,区分人乳头瘤病毒(hpv)18型dna和rna模板
[0079]
本实施例从宫颈脱落细胞中分别提取dna和rna,并在相同条件下分别进行一步法rt-pcr,最终利用毛细管电泳分离样品,检验本发明的引物对dna和rna模板的区分能力,具体步骤如下:
[0080]
(1)根据图2中的方案设计hpv 18型e6/e7基因的反转录(反向)引物,并根据常规引物设计方案设计正向引物,并交由上海生工生物有限公司进行引物合成,其中正向引物5’端标记fam基团。具体的正向引物序列为5
’-
tatgctgcaaccgagcacga-3’(seq id no:3),
反转录(反向)引物序列为5
’-
gatggacggacagttctgctgagctttcta-3’(seq id no:4),其中小写字母部分为人为引入的错配碱基,大小字母部分为与rna模板互补部分,目标产物大小为323bp。
[0081]
(2)获取确诊为hpv 18型感染患者的宫颈脱落细胞样本,并通过dna提取试剂盒和rna提取试剂盒分别对样本进行dna和rna提取,具体操作方法详见市售基因组dna提取试剂盒与rna提取试剂盒操作说明。需要注意最终用于一步法rt-pcr的dna样本需经过rnase a处理以去除rna残留,rna样本需经过dnase i处理以去除dna残留,并对dna和rna模板样本进行精确定量。
[0082]
(3)在pcr八联管中分别加入dna和rna模板各2μl(浓度为1e+3拷贝/μl),rt-pcr酶0.5μl,上述正反向引物(1μm)各0.5μl,2
×
pcr缓冲液5μl,dutp(100mm)0.01μl,ddh2o补足反应体系10μl。
[0083]
(4)将配制好的体系离心混匀,按表2程序进行rt-pcr反应。
[0084]
表2 一步法rt-pcr反应程序
[0085][0086]
(5)准备电泳样品,每个电泳孔分别加入hi-di 8.7μl,size-5000.3μl,pcr产物1μl,混匀后离心备用。
[0087]
(6)毛细管电泳分离样品,将样品板置于3500dx基因分析仪中,选择“fragment”电泳方法,进行电泳,具体操作方法详见abi3500操作说明书。
[0088]
(7)结果分析。如图20所示,以提取的rna为模板的一步法rt-pcr毛细管电泳结果显示,一步法rt-pcr扩增出了片段长度为323bp的pcr产物,与设计引物时的目的片段大小一致。而图21则表明,以相同浓度dna为模板的一步法rt-pcr毛细管电泳结果显示,一步法rt-pcr并没有扩增出323bp的产物。由此可知,通过本发明设计的引物能够在一步法rt-pcr过程中实现对rna和dna的选择性扩增,能够很好的区分rna和dna模板。
[0089]
实施例3:利用设计的引物进行一步法rt-pcr,区分粪肠球菌(enterococcus faecalis)细胞溶素编码基因dna和rna模板
[0090]
酒精性肝炎是一种由酒精过度使用引起的严重危及生命的肝病,最新研究表明,酒精性肝炎患者粪便中的粪肠球菌数量远远大于非酒精性肝炎患者,且粪肠球菌分泌的细胞溶素与酒精性肝炎疾病严重程度和死亡率呈正相关性,若能直接在粪便样品中检测粪肠
球菌细胞溶素编码基因的表达情况,就能够更好的表征患者酒精性肝炎的严重程度。
[0091]
本实施例从粪便样本中分别提取dna和rna,并在相同条件下分别进行一步法rt-pcr,最终利用毛细管电泳分离样品,检验本发明的引物对dna和rna模板的区分能力,具体步骤如下:
[0092]
(1)根据图5中的方案设计针对粪肠球菌细胞溶素编码基因(cyls)的反转录(反向)引物,并根据常规引物设计方案设计正向引物,并交由上海生工生物有限公司进行引物合成,其中正向引物5’端标记fam荧光基团。具体的正向引物序列为5
′-
aaattagaacttgttggtcct-3
′
(seq id no:5),反转录(反向)引物序列为5
’-
ccatggactattttttttttttttacctactcctaagcc-3’(seq id no:6),其中小写字母碱基部分为人为引入的非连续错配碱基,带下划线的t碱基部分为连接用碱基,大写字母碱基部分为与rna模板互补部分,产物大小为150bp。
[0093]
(2)同时采用常规引物设计方案,以人类β-actin作为反应内参进行引物设计,其正向引物序列为:5
’-
gcggactatgacttagttgcgttacaccct-3’(seq id no:7),5’端标记fam荧光基团;其反向引物为5
’-
ggggatgctcgctccaaccg-3’(seq id no:8),产物长度为191bp。
[0094]
(3)获取确诊为酒精性肝炎患者和非酒精性肝炎患者粪便样本,分别利用粪便dna提取试剂盒和粪便rna提取试剂盒对粪便dna和rna进行提取,具体操作方法详见市售粪便dna、rna提取试剂盒操作说明书。提取完成后将每份rna样本分成两份,其中一份rna样本进行dna酶消化处理,其中一份rna样本不做任何处理,最终共获得2份dna样本和4份rna样本,分别为酒精性肝炎患者dna样本(简称dna样本3-1)、非酒精性肝炎患者dna样本(简称dna样本3-2)、未经dna酶消化的酒精性肝炎患者的rna样本(简称rna样本3-1)、经过dna酶消化的酒精性肝炎患者的rna样本(简称rna样本3-2)、未经dna酶消化的非酒精性肝炎患者的rna样本(简称rna样本3-3)、经过dna酶消化的非酒精性肝炎患者的rna样本(简称rna样本3-4),并对各dna和rna样本进行精确定量,-80℃保存备用。
[0095]
(4)在四个pcr管中分别加入相同浓度的rna样本
[0096]
3-1/3-2/3-3/3-4各2μl,rt-pcr酶0.5μl,各个正反向引物(1μm)0.5μl,2
×
pcr buffer 5μl,ddh2o补足反应体系10μl,体系离心混匀后使用一步法反转录pcr程序进行扩增(表3)。
[0097]
表3 一步法rt-pcr反应程序
[0098][0099]
(5)另外,在其他四个pcr管中分别加入相同浓度的rna样本3-1/3-2/3-3/3-4各2μl,rtaq酶0.5μl,各个正反向引物(1μm)0.5pl,2
×
pcr buffer 5μl,ddh2o补足反应体系10μl,体系离心混匀后使用常规pcr程序进行扩增(表4)。
[0100]
表4常规pcr反应程序
[0101][0102]
(6)另外,在其他两个pcr管中分别加入相同浓度的dna样本3-1/3-2各2μl,rtaq酶0.5μl,各个正反向引物(1μm)0.5μl,2
×
pcr buffer 5μl,ddh2o补足反应体系10μl,体系离心混匀后同样使用常规pcr程序进行扩增(表4)。
[0103]
(7)准备电泳样品,每个电泳孔分别加入hi-di 8.7μl,size-5000.3μl,pcr产物1μl,混匀后离心备用。
[0104]
(8)毛细管电泳分离样品,将样品板置于3500dx基因分析仪中,选择“fragment”电泳方法,进行电泳,具体操作方法详见abi3500操作说明书。
[0105]
(9)结果分析:rna样本3-1和3-2的rt-pcr与常规pcr的cyls的检出峰的比较(参见图22-25)表明,残留dna无法在pcr过程中与反向引物进行较好的退火,所以无法进行有效的扩增,而一旦经过了反转录步骤,则将人为引入的错配碱基加入到了pcr的模板中,进而能够在pcr过程中非常完美的退火,形成pcr产物并被检出(图22和23),且数据表明rna样本3-1在rt-pcr中的荧光信号全部来自rna模板,而非部分来自于残留的dna模板(图23中,存在残留的dna,仍未检出目标产物)。再比较β-actin检出峰:对于相同的程序,rna样本3-1的β-actin检出峰高于rna样本3-2的β-actin检出峰,这是因为残留的dna也可作为β-actin扩
增的模板并且经过dna酶消化后rna模板部分降解(参见图22和24;图23和25)。rna样本3-1和3-2的rt-pcr的cyls的检出峰(图22和24)比较表明,由于dna酶消化的过程中rna出现部分降解,导致rna样本cyls荧光信号降低。rna样本3-3和3-4(图26-29)的β-actin检出峰表现出和rna样本3-1和3-2相同的变化趋势,分析同上。dna样本3-1在rt-pcr和pcr过程(图30和31)中均未检出cyls,进一步说明本发明的引物设计方法具有很强的rna模板识别能力,能够很好的区别dna和rna模板。上述结果表明,通过反转录引物设计方式4进行反转录引物设计,能够很好的区分dna和rna模板,且能够很好的减少dna酶消化过程中rna的降解,提高检测的灵敏度和准确性。
[0106]
实施例4:利用设计的引物进行一步法rt-pcr,精准检测乙肝病毒dna多聚酶/逆转录酶的mrna表达
[0107]
乙型肝炎病毒(hepatitis b virus,hbv)感染是一种严重影响公共卫生与人体健康的全球性流行疾病,其在人体中复制主要包括如下步骤:
①
黏附:hbv表面抗原(hbsag)识别肝细胞并实现黏附;
②
脱壳:进入肝细胞并脱去核心抗原(hbcag)和e抗原(hbeag),同时暴露出hbv核酸;
③
入核:从肝细胞浆进入肝细胞核,形成共价闭合环状脱氧核糖核酸(cccdna);
④
转录:借助人体酶系将共价闭合环状脱氧核糖核酸(cccdna)转录成mrna;
⑤
翻译:将mrna翻译成蛋白质,包括hbv外膜蛋白(hbsag)、hbv核壳蛋白(hbeag、hbcag)、hbv dna多聚酶/逆转录酶(rt)等;
⑥
逆转录:利用翻译生成的hbv dna多聚酶/逆转录酶(rt)将hbv mrna逆转录成hbv dna;
⑦
组装:将所有合成的hbv蛋白质和dna组成并释放到健康肝细胞或血液中。
[0108]
由此可见,在这个复制过程中最为关键的是hbv dna多聚酶/逆转录酶(rt)的形成,若该酶无法形成,乙肝病毒就无法完成逆转录过程,生成hbv dna,也就无法完成组装和释放过程,且研究表明hbv dna多聚酶/逆转录酶的活跃表达与乙型肝炎的慢性化和慢性乙型肝炎迁延不愈有直接联系。
[0109]
本实施例从血液样本中分别提取dna和rna,并在相同条件下分别进行一步法rt-pcr,最终利用毛细管电泳分离样品,检验本发明的引物对dna和rna模板的区分能力,具体步骤如下:
[0110]
(1)根据图9中的方案设计针对hbv多聚酶/逆转录酶编码基因的反转录(反向)引物,并根据常规引物设计方案设计正向引物,并交由上海生工生物有限公司进行引物合成,其中正向引物5’端标记fam基团。具体的正向引物序列为
[0111]5’-
ctggatgtgtctgcggcgttttatc-3’(seq id no:9),反转录(反向)引物序列为
[0112]5’-
ctatcatctccttttttttttcaaacggtcgacata-3’(seq id no:10),其中小写字母碱基部分为人为引入的非连续错配碱基,带下划线的t碱基部分为连接用碱基,大写字母碱基部分为与rna模板互补部分,产物大小为125bp。
[0113]
(2)同时采用常规引物设计方案,以人类β-actin作为反应内参进行引物设计,其正向引物序列为:
[0114]5’-
gagcgagcatcccccaaagttcacaa-3’(seq id no:11),5’端标记fam荧光基团;其反向引物为
[0115]5’-
ctccttagagagaagtggggtggcttttagg-3’(seq id no:12),产物长度为145bp。
[0116]
(3)获取确诊为乙肝病毒携带且hbv dna检测为中量复制的患者和无乙肝病毒携
带者的血液样本,分别利用市售血液rna提取试剂盒和血液dna提取试剂盒进行rna和dna提取,具体操作方法详见操作说明说明书。提取完成后将每份rna样本分成两份,其中一份rna样本进行dna酶消化处理,另一份rna样本不做任何处理,最终获得4份rna样本和2份dna样本,分别为未经dna酶消化的乙肝病毒携带者的rna样本(简称为rna样本4-1)、经过dna酶消化的乙肝病毒携带者的rna样本(简称为rna样本4-2)、未经dna酶消化的无乙肝病毒携带者的rna样本(简称为rna样本4-3)、经过dna酶消化的无乙肝病毒携带者的rna样本(简称为rna样本4-4)、乙肝病毒携带者的dna样本(简称dna样本4-1)以及非乙肝病毒携带者的dna样本(简称dna样本4-2),并对dna和rna模板样本进行精确定量,-80℃保存备用。
[0117]
(4)在四个pcr管中分别加入相同浓度的rna样本
[0118]
4-1/4-2/4-3/4-4各2μl,rt-pcr酶0.5μl,各个正反向引物(1μm)0.5μl,2
×
pcr buffer5μl,ddh2o补足反应体系10μl,体系离心混匀后使用一步法反转录pcr程序进行扩增(表5)。
[0119]
表5 一步法rt-pcr反应程序
[0120][0121]
(5)另外,在其他四个pcr管中分别加入相同浓度的rna样本4-1/4-2/4-3/4-4各2μl,rtaq酶0.5μl,各个正反向引物(1μm)0.5μl,2
×
pcr buffer 5μl,ddh2o补足反应体系10μl,体系离心混匀后使用常规pcr程序进行扩增(表6)。
[0122]
表6常规pcr反应程序
[0123][0124]
(6)另外,在其他两个pcr管中分别加入相同浓度的dna样本4-1/4-2各2μl,rtaq酶
0.5μl,各个正反向引物(1μm)0.5μl,2
×
pcr buffer 5μl,ddh2o补足反应体系10μl,体系离心混匀后同样使用常规pcr程序进行扩增(表6)。
[0125]
(7)准备电泳样品,每个电泳孔分别加入hi-di 8.7μl,size-5000.3μl,pcr产物1μl,混匀后离心备用。
[0126]
(8)毛细管电泳分离样品,将样品板置于3500dx基因分析仪中,选择“fragment”电泳方法,进行电泳,具体操作方法详见abi3500操作说明书。
[0127]
(9)结果分析:rna样本4-1和4-2的rt-pcr与pcr(参见图34-37)的hbv检出峰的比较表明,残留的dna无法在pcr过程中与反向引物进行有效退火,所以无法进行扩增,而一旦经过了反转录步骤,则将人为引入的错配碱基加入到了pcr的模板中,进而能够在pcr过程中非常完美的退火,形成pcr产物并被检出(图34和35),且表明rna样本4-1在rt-pcr中的荧光信号全部来自于rna模板,而非部分来自于残留的dna(图35中,存在残留的dna,仍未检出目标产物)。再比较β-actin检出峰:对于相同的程序,rna样本4-1的β-actin检出峰高于rna样本4-2的β-actin检出峰,这是因为残留的dna也可作为β-actin扩增的模板并且经过dna酶消化后rna模板部分降解(参见图34和36;图35和37)。rna样本4-1和4-2的rt-pcr hbv检出峰(图34和36)的比较表明,由于dna酶消化过程中rna出现了部分降解,导致hbv荧光信号降低。rna样本4-3和4-4(图38-41)的β-actin检出峰表现出和rna样本4-1和4-2相同的变化趋势,分析同上。dna样本4-1(图42和43)在rt-pcr和pcr过程中均未检出hbv,进一步说明本发明的引物设计方法具有很强的rna模板识别能力,能够很好的区别dna和rna模板。上述结果表明,通过反转录引物设计方式8进行反转录引物设计,能够很好的区分dna和rna模板,且能够很好的减少dna酶消化过程中rna的降解,提高了检测的灵敏度和准确度。
[0128]
实施例5:利用设计的引物/探针进行反转录荧光定量pcr
[0129]
(rt-qpcr)区分人乳头瘤病毒(hpv)45型dna和rna模板
[0130]
本实施例从宫颈脱落细胞中分别提取dna和rna,并在相同条件下分别进行rt-qpcr,检验本发明的引物对dna和rna模板的区分能力,具体步骤如下:
[0131]
(1)根据图13中的方案设计hpv 45型e6/e7基因的反转录(反向)引物,并根据常规引物设计方案设计正向引物,并交由上海生工生物有限公司进行引物合成。具体的正向引物序列为5
’-
ttgtgttacgagcaattaagcgagtcag-3’(seq id no:13),反转录(反向)引物序列为
[0132][0133]
(seq id no:14),其中小写字母且无下划线部分为人为引入的错配碱基,大写字母且有下划线部分为连接用碱基,小写字母且有下划波浪线部分为保护碱基和保护碱基互补部分,大写字母部分为与rna模板互补部分,目标产物大小为125bp。同时设计探针,在探针5’端标记fam荧光基团,3’端标记bhq-1荧光猝灭基团,同样交由上海生工生物有限公司进行探针合成。具体的探针序列为5
’-
tcatgcacaactaccagcccgacgag-3’(seq id no:15)。
[0134]
(2)同时采用常规引物设计方案,以人类β-actin作为反应内参进行引物设计,其正向引物序列为:5
’-
gcatccacgaaactaccttcaactcca-3’(seq id no:16),其反向引物为5
’-
ccgccagacagcactgtgtt-3’(seq id no:17),产物长度为88bp。同时设计探针,在探针5’端标记vic荧光基团,3’端标记bhq-2荧光猝灭基团,同样交由上海生工生物有限公司进行探针合成。具体的探针序列为5
’-
aagtgtgacgtggacatccgcaaagacc-3’(seq id no:18)。
[0135]
(3)获取确诊为hpv 45型感染患者的宫颈脱落细胞样本,并通过dna提取试剂盒和rna提取试剂盒分别对样本进行dna和rna提取,具体操作方法详见市售基因组dna提取试剂盒与rna提取试剂盒操作说明。需要注意dna样本需经过rnase a处理以去除rna残留,rna样本需经过dnase i处理以去除dna残留,并对dna模板样本(简称为dna样本5-1)和rna模板样本(简称为rna样本5-1)进行精确定量。
[0136]
(4)在不同pcr管中分别加入相同浓度的rna样本5-1和dna样本5-1各5μl,rt-pcr酶1μl,各个正反向引物(10μm)0.5μl,探针(10μm)0.2μl,2
×
pcr buffer 10μl,ddh2o补足反应体系20μl,体系离心混匀后使用rt-qpcr程序进行扩增(表7)。
[0137]
表7多重反转录实时荧光定量pcr反应程序
[0138][0139]
(5)结果分析:rna样本5-1和dna样本5-1的rt-qpcr扩增曲线比较表明,由于dna样本在pcr过程中无法与反向引物进行有效的退火,所以无法进行扩增,故在fam通道无荧光信号检出,而rna样本经过了rt过程,将人为引入的错配碱基加入到了pcr模板中,进而能够在pcr过程中完美退火,形成pcr产物,并在fam通道中检出荧光信号,表明本发明的引物设计方式能够很好的区分dna和rna模板(图46和48)。另外vic通道扩增曲线比较表明,常规引物设计方式无法区分dna和rna模板,均在通道中检出了荧光信号(图47和49),进一步证明本发明的引物设计方式对于dna和rna模板的特异性识别功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1