一种1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的制作方法
1.本发明属于有机药物共晶技术领域,特别涉及一种1,2-二(4-吡啶基)乙烷-阿昔莫司共晶。
背景技术:
2.药物共晶是基于超分子化学原理,即通过分子间的相互协同作用进行的分子识别和超分子自组装。药物活性成分(api)与合适的共晶形成物(cocrystal former,ccf)通过氢键自组装,或者带有饱和性和方向性的非共价键(如芳烃或苯环的范德华力,π-π共轭作用和卤键)组装形成的一种新型结构,即药物共晶。它以氢键为基础,既不需要形成新的共价键,又不需要破坏已有的共价键,在保留药物本身的药理作用的同时,又能修饰药物的理化性质,如提高药物的稳定性、降低其引湿性、提高溶解性、提高生物利用度等,为药物共晶在制药工业方面的应用提供了广阔的发展前景。近几年来,药物共晶研究越来越受到人们的关注。现阶段,国外对药物共晶的研究开始逐渐增多并深入;而国内对其研究还相对较少。对于仿制药来说,药物共晶的研究也可以打破原研药公司对药物晶型的专利保护,利于将仿制药推向市场。因此,获得更多具有新颖、实用和创造性的药物共晶具有重要的现实意义,特别是一些水不溶性药物。
3.阿昔莫司为烟酸衍生物,是一种广谱长效调血脂药,用于各种原发性和继发性高脂血症,主要作用于脂肪组织,通过抑制脂肪组织释放游离脂肪酸,减少血浆低密度脂蛋白及极低密度脂蛋白的合成,从而降低血浆中血浆低密度脂蛋白及极低密度脂蛋白的水平,同时通过抑制肝脂肪酶活性而增高血浆hdl水平。阿昔莫司由意大利farmitalia carlo erba公司研制开发,于1985年在意大利上市,而后,凭借其较高的安全性及显著的疗效,相继在德国、智利、瑞士、中国香港等多个国家和地区上市。
4.药物共晶会影响药物的理化性质,直接影响药物在生理ph7.4条件下的溶出及吸收效率,进而影响药物的生物利用度,临床疗效等。通过药物共晶的方式,可以很好的应用共晶优势,这对于理解和掌握药物有效分子的空间排布以及理化性质具有非常重要的作用。
5.目前关于阿昔莫司的相关报道较多,但是主要是关于其制备、制剂、理化性质及药理等性质的报道,关于其晶体共晶结构的报道较少,专利us2005239803a1、cn 103508963 a等均报道了阿昔莫司的制备方法,专利cn86103304-2得到晶体性状的阿昔莫司沉淀物,为阿昔莫司水合物,产率较低。在之前的报道中,对于阿昔莫司晶体共晶报道较少,阿昔莫司共晶晶体学表征参数未有提及。
技术实现要素:
6.鉴于现有技术的不足,本发明提供了一种1,2-二(4-吡啶基)乙烷-阿昔莫司共晶。
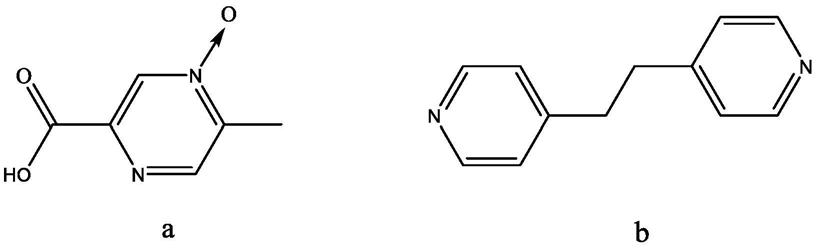
7.阿昔莫司作为本发明的药物成分,化学名为5-甲基吡嗪-2-羧酸-4-氧化物,为白色或类白色结晶性粉末。cas号:51037-30-0,分子式为c6h6n2o3,其结构式如a所示,发明中
所选择的共晶形成物为1,2-二(4-吡啶基)乙烷,分子式为c
12
h
12
n2,其结构式如b所示。
[0008][0009]
本发明的第一方面,提供了1,2-二(4-吡啶基)乙烷-阿昔莫司共晶。所述共晶体中,阿昔莫司与1,2-二(4-吡啶基)乙烷的摩尔比为2:1。
[0010]
所述的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶,使用cu-kα辐射,以2θ表示的x射线衍射谱图在9.88
±
0.2
°
,13.92
±
0.2
°
,24.56
±
0.2
°
,26.12
±
0.2
°
,26.86
±
0.2
°
有特征峰。
[0011]
优选地,所述的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶,使用cu-kα辐射,以2θ表示的x射线衍射谱图在9.88
±
0.2
°
,13.92
±
0.2
°
,15.38
±
0.2
°
,15.70
±
0.2
°
,24.56
±
0.2
°
,26.12
±
0.2
°
,26.86
±
0.2
°
,29.59
±
0.2
°
,36.90
±
0.2
°
有特征峰。
[0012]
优选地,所述的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶,使用cu-kα辐射,其特征峰符合如图1所示的x射线粉末衍射图谱。
[0013]
优选地,所述的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶,其在差示扫描量热曲线dsc中存在吸热峰,为184.43℃。
[0014]
优选地,所述的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶,其晶体学参数是:其晶体学参数是:单斜晶系,空间群为p2
1/c
;晶胞参数为:α=90
°
,β=99.3338(11)
°
,γ=90
°
,晶胞体积
[0015]
本发明的第二方面提供一种1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的制备方法,具体制备步骤包括:将阿昔莫司和1,2-二(4-吡啶基)乙烷加入有机溶剂a中,加热溶解,溶液澄清后,控制温度至回流,反应,降温析晶,过滤干燥得1,2-二(4-吡啶基)乙烷-阿昔莫司共晶。
[0016]
所述的有机溶剂a选自甲醇、乙腈、丙酮、乙酸乙酯、四氢呋喃、乙醇、异丙醇的一种或两种。
[0017]
优选地,所述有机溶剂a选自甲醇、乙醇、乙酸乙酯的一种或两种。
[0018]
所述阿昔莫司与1,2-二(4-吡啶基)乙烷的摩尔比为1:0.4~0.6。
[0019]
优选地,阿昔莫司与1,2-二(4-吡啶基)乙烷的摩尔比为1:0.5。
[0020]
所述体系中1,2-二(4-吡啶基)乙烷和有机溶剂a的质量体积比为4~6:1,其中质量以mg计,体积以ml计。
[0021]
所述的降温析晶温度为0~10℃。
[0022]
优选地,降温析晶温度为5~10℃。
[0023]
所述的析晶时间为30~50小时。
[0024]
进一步优选地,所述制备方法包括以下步骤:
[0025]
将阿昔莫司和1,2-二(4-吡啶基)乙烷溶于有机溶剂a中,50~80℃加热溶解,搅拌回流反应7-10小时,降温至0~10℃析晶30~50小时,过滤,洗涤滤饼,干燥得1,2-二(4-吡啶基)乙烷-阿昔莫司共晶。
[0026]
所述洗涤滤饼的溶剂选自甲醇、乙醇、乙酸乙酯中的一种。
[0027]
所述干燥温度为55~75℃,干燥时间为8~10小时。
[0028]
本发明的第三方面提供一种1,2-二(4-吡啶基)乙烷-阿昔莫司共晶作为活性成分制备治疗降血脂药中的用途。
[0029]
晶体结构的确认
[0030]
x射线晶体数据在日本理学xtalab synergy型号仪器上收集,测试温度293(2)k,用cuka辐射,以ω扫描方式收集数据并进行lp校正。晶体结构用olex2软件中的shelxt程序计算得到,并采用shelxl程序通过最小二乘法修正结构参数和判别原子种类,使用几何计算法和差值fourier法获得全部氢原子位置,拟合优度(goof值)1.123,接近于1.0,表明权重方案合适、结构准确。
[0031]
测试及解析本发明制备的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶所得晶体学数据是(表1):其晶体学参数是:单斜晶系,空间群为p2
1/c
;晶胞参数为:;晶胞参数为:α=90
°
,β=99.3338(11)
°
,γ=90
°
,晶胞体积分子式是:c
24
h
24
n6o6,分子量是:492.49。本发明的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶晶体的ortep图表明,阿昔莫司和1,2-二(4-吡啶基)乙烷通过分子内氢键连接在一起,其中阿昔莫司羧基h4与1,2-二(4-吡啶基)乙烷上的n4形成氢键,另一个阿昔莫司分子羧基上h3与1,2-二(4-吡啶基)乙烷上的n3形成分子内氢键,如附图3所示。本发明的1,2-二(4-吡啶基)乙烷-阿昔莫司的堆积图,如附图2所示。
[0032]
表1 1,2-二(4-吡啶基)乙烷-阿昔莫司共晶主要晶体学数据
[0033][0034]
x-射线粉末衍射测试仪器及测试条件:x-射线粉末衍射仪:panalyticale;cu-kα;样品台:平板;入射光路:bbhd;衍射光路:plxcel;电压45kv,电流40ma;发散狭缝:1/4;防散射狭缝:1;索拉狭缝:0.04rad;步长:0.5s;扫描范围:3~50
°
。
[0035]
依据上述晶体学数据,其对应的x射线粉末衍射图(cu-kα)中特征峰详见附图1及表2。
[0036]
表2 1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的pxrd峰
[0037]
[0038][0039]
实施例中所制备的所有样品都具有相同的晶体学参数及x射线粉末衍射谱图。
[0040]
本发明所述方法制备的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶tga/dsc测试结果如图4所示,dsc检测结果有一个吸热峰,对应温度为184.43℃。根据tga检测结果可以看出存在一个失重台阶,结合dsc/tga检测结果表明,本发明制备的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶不含其它溶剂。
[0041]
本发明所述方法制备的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶相对于目前报道的阿昔莫司晶型具有以下优势:
[0042]
稳定性高,1,2-二(4-吡啶基)乙烷-阿昔莫司共晶经过光照试验和高温、高湿环境放置后,hplc纯度仍然高于99.70%,其固体状态稳定性好。
附图说明
[0043]
图1:1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的x射线粉末衍射图谱。
[0044]
图2:1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的堆积图。
[0045]
图3:1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的ortep图。
[0046]
图4:1,2-二(4-吡啶基)乙烷-阿昔莫司共晶的差示扫描量热曲线(dsc)图。
具体实施方式
[0047]
现通过以下实施例来进一步描述本发明的有益效果,实施例仅用于例证的目的,不限制本发明的范围,同时本领域普通技术人员根据本发明所做的显而易见的改变和修饰也包含在本发明范围之内,杂质i为5-甲基吡嗪-2-羧酸。
[0048]
实施例1
[0049]
将154.12mg阿昔莫司,92.12mg1,2-二(4-吡啶基)乙烷加入到20ml的甲醇中,搅拌溶解,控温65℃,回流反应8h,反应完毕,缓慢降温至5~10℃后,静置析晶40h,析晶结束,过滤,甲醇洗涤滤饼,50℃下真空干燥12h,得1,2-二(4-吡啶基)乙烷阿昔莫司共晶,收率98.33%,纯度99.95%。
[0050]
实施例2
[0051]
将154.12mg阿昔莫司,73.70mg1,2-二(4-吡啶基)乙烷加入到15ml乙醇中,搅拌溶解,控温至回流,反应10h,反应完毕,缓慢降温至0~5℃后,静置析晶40h,析晶结束,过滤,乙醇洗涤滤饼,55℃下真空干燥8h,得1,2-二(4-吡啶基)乙烷阿昔莫司共晶,收率96.56%,纯度99.94%。
[0052]
实施例3
[0053]
将154.12mg阿昔莫司,110.54mg1,2-二(4-吡啶基)乙烷加入到25ml乙酸乙酯中,搅拌溶解,控温至回流,反应7h,反应完毕,缓慢降温至10~15℃后,静置析晶48h,过滤,甲醇洗涤滤饼,60℃下真空干燥10h,得1,2-二(4-吡啶基)乙烷阿昔莫司共晶,收率96.88%,纯度99.90%。
[0054]
实施例4
[0055]
将154.12mg阿昔莫司,64.48mg1,2-二(4-吡啶基)乙烷加入到10ml的丙酮中,搅拌溶解,控温至回流,反应8h,反应完毕,缓慢降温至-5~0℃后,静置析晶35h,过滤,用乙酸乙酯洗涤滤饼,50℃下真空干燥10h得1,2-二(4-吡啶基)乙烷阿昔莫司共晶,收率94.51%,纯度99.82%。
[0056]
实施例5
[0057]
将154.12mg阿昔莫司,119.76mg1,2-二(4-吡啶基)乙烷加入到35ml的乙腈中,搅拌溶解,控温至回流,反应7h,反应完毕,缓慢降温至5~10℃后,静置析晶30h,过滤,用甲醇洗涤滤饼,50℃下真空干燥12h,得1,2-二(4-吡啶基)乙烷阿昔莫司共晶,收率94.33%,纯度99.84%。
[0058]
实施例6
[0059]
将154.12mg阿昔莫司,92.12mg1,2-二(4-吡啶基)乙烷加入到20ml混合溶剂(10ml甲醇+10ml乙醇)中,控温至回流,反应8h,反应完毕,缓慢降温至5~10℃后,控温静置析晶40h,过滤,用甲醇洗涤滤饼,50℃下真空干燥10h,得1,2-二(4-吡啶基)乙烷阿昔莫司共晶,收率97.12%,纯度99.90%。
[0060]
对比实施例1:
[0061]
在10l玻璃反应釜中加入2730ml质量浓度为98%的浓硫酸,搅拌条件下加入910.0g的5-甲基吡嗪-2,3-二羧酸,加热至60℃,加热反应1h,然后缓慢加入5.5kg水、164.9g钨酸钠(na2wo4·
2h2o),623.0g质量浓度为30%的双氧水,继续加热搅拌8h,冰浴条件下冷却析晶4h,抽滤固体,100℃下干燥12h,制备出产品阿昔莫司595g。该反应中产品收率77.30%;hplc纯度96.20%,杂质i:2.80%。
[0062]
对比实施例2:
[0063]
向100g阿昔莫司粗品中加入200ml水,加热至100℃,搅拌溶解后加入3.0g活性炭继续保温搅拌20分钟,抽滤;将滤液以10℃/h降温至60℃,然后向其中滴加220g丙酮,滴毕,以10℃/h降温至5℃析晶7h,抽滤,用丙酮洗涤滤饼,烘干(0.01mpa,80℃)即得类白色的阿昔莫司,收率为88.60%。hplc纯度:98.30%,5-甲基吡嗪-2-羧酸(杂质i):0.50%。
[0064]
对比实施例3:
[0065]
将330mg(1mmol)的na2wo4·
2h2o置于50ml烧瓶中,以16ml水溶解并配以机械搅拌、回流冷却器及温度计。将3.75ml 40%重量/体积(400g/l)(44mmol)的过氧化氢加入于溶液中,用稀h2so4调至ph值为1.5,然后加入5.52g(40mmol)的2-羧基-5-甲基吡嗪。
[0066]
反应产生的水的悬浮物在搅拌下加热至70℃并维持在这温度2.5小时。因而得到逐渐增溶的悬浮物。最后发现有部分产物沉淀。将混合物在室温下静置过夜,而产生晶体形状的反应产物的沉淀。这产物经过滤及用冰水洗涤,再置于素烧板上干燥便可获得部分是水合式(2.83%)的2-羧基-5-甲基吡嗪-4-氧化物4.68g,相当于4.54g的无水产物。产率为73.01%。hplc纯度:95.10%,杂质i:2.30%。
[0067]
对比实施例4:
[0068]
将250mg(0.75mg)的na2wo4·
2h2o置于50ml烧瓶中,以13ml水溶解并配以机械搅拌、回流冷却器及温度计。将3.23ml40%重量/体积(400g/l)(38mmol)的过氧化氢加入于溶液中,用稀h2so4调至ph值为2.0,然后加入3.76g 98%(30mmol)的2-羧基-5-甲基吡嗪。
[0069]
反应产生的水的悬浮物在搅拌下加热至80℃并维持在这温度2小时。而45min后即可获得完全增溶的悬浮物。最后,溶液在室温下静置过夜而产生晶体形状的反应产物的沉淀。这产物经过滤及用冰水洗涤,再置于素烧板上干燥获得3.02g 2-羧基-5-甲基吡嗪-4-氧化物的一水合物(实验值h2o-11.35%;一水合物产物的计算值h2o-11.3%),产率为63.02%。hplc纯度:94.21%,杂质i:3.40%。
[0070]
对比实施例5:
[0071]
将2-羧基-5-甲基吡嗪4-氧化物(2.5g)加入到甲醇(60ml)和乙醇胺(1.1ml)的混合溶液中。混合物加热回流20分钟,然后冷却并过滤,从甲醇结晶后得到2-羧基-5-甲基吡嗪4-氧化物乙醇胺盐(2.1g),mp.177
°-
180℃,收率:60.17%,hplc纯度:96.81%,杂质i:2.10%。
[0072]
对比实施例6:
[0073]
氮气保护下,在配有机械搅拌器、水冷凝器(带有气体入口)和热电偶的500ml.x.4颈瓶中进行反应。向反应器中加入三甲基硅醇钠(3.71g)和thf(90g),然后加入5-甲基吡嗪羧酸-4-氧化物乙酯(6.00g),将混合物在室温下搅拌4小时,过滤收集固体并用thf(3x45g)冲洗。真空干燥(25英寸汞柱,65℃),得到5.38g(收率:92.50%)钠盐,为灰白色固体,hplc纯度:96.80%,杂质i:2.41%。
[0074]
稳定性试验
[0075]
温湿度及光照试验
[0076]
具体的稳定性试验方法参照中国药典2015版第四部有关稳定性考察的指导方法进行,纯度检测用hplc法进行检测,具体的试验结果见下表。
[0077]
表3甲基吡嗪衍生物晶型在光照、高温及高湿条件下的稳定性试验结果
[0078]
[0079][0080]
经试验,本发明制备得到所有的1,2-二(4-吡啶基)乙烷甲基-阿昔莫司共晶均可达相近的稳定性效果。本发明制备得到的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶在光照、高温及高湿的条件下其纯度、外观均未发生比较明显的变化,而对比例1至对比例6晶型在相同的实验条件下其纯度大幅降低,其杂质含量都有较明显的升高,即出现了变质的情况,可见本发明制备的1,2-二(4-吡啶基)乙烷-阿昔莫司共晶相比于现有的晶型具有较好的化学稳定性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1