利伐沙班噻吩羧酸酯杂质对照品及其制备方法与流程
本发明涉及药物杂质,具体涉及一种利伐沙班噻吩羧酸酯杂质对照品及其制备方法。
背景技术:
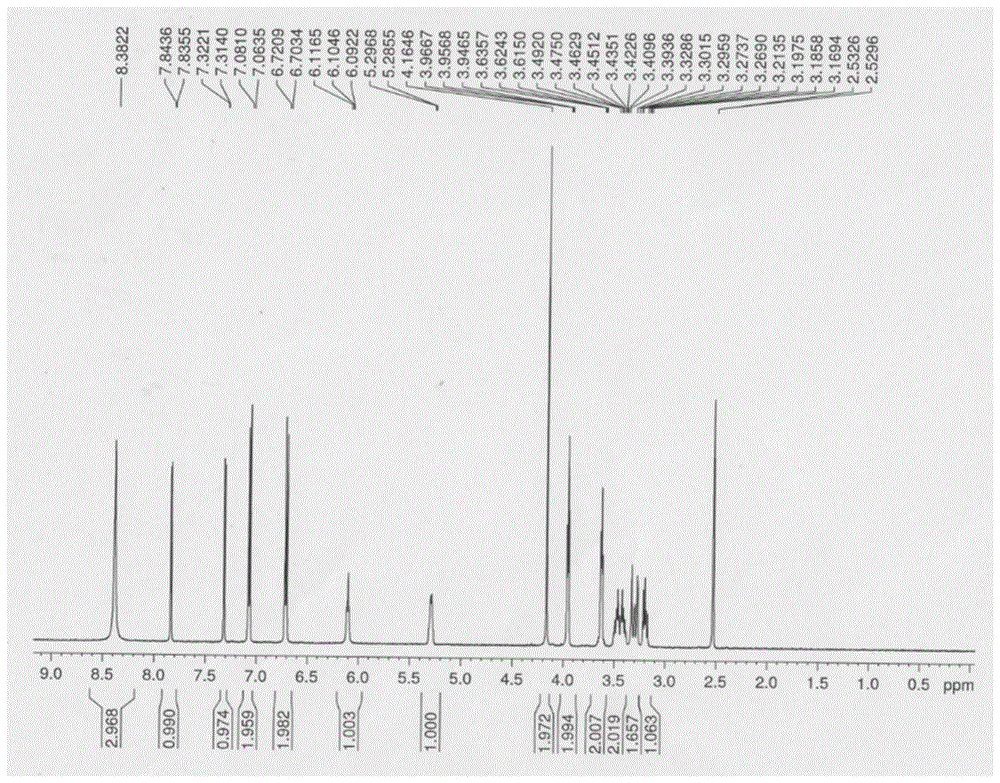
:利伐沙班(rivaroxaban,结构式如式ii所示)是全球第一个高选择性直接抑制因子xa的口服抗凝药,是拜耳公司历经10年研发的新型抗凝药物,为化学合成的小分子化合物,并且是唯一的一种疗效始终优于依诺肝素的新型口服抗凝药,主要用于预防髋关节和膝关节置换术后患者深静脉血栓(dvt)和肺栓塞(pe)的形成。在药物工艺研发过程中,杂质作为药品的一项关键质量属性,是影响药物疗效和安全性的一个重要因素。因此对药物杂质进行全方位研究并进行有效控制对于确保药物质量意义重大。当前药物杂质研究已经从最初的药物纯度控制经由单一杂质的控制上升到到杂质谱的研究高度,这一发展过程也越发突显了药物杂质研究对药物开发的重要性。杂质谱是对存在于药品中所有已知杂质和未知杂质的总的描述,包括已鉴定杂质(即已确证了结构特征的杂质),特定杂质(即在质量标准中规定检查并有自己限度标准的已鉴定或未鉴定的杂质)以及潜在杂质(即理论推测在生产或贮藏过程中可能产生的,实际产品中不一定存在的杂质)。只有在全面杂质谱分析基础上,药品质量控制才能得到有效保障。药品上市前必须进行质量、安全性和效能科学评价,药物杂质与药物质量以及药物安全性密切相关,杂质对照品可帮助确定杂质的合理限度,对药品检验方法及质量标准的建立将会起着极大的促进作用,利伐沙班噻吩羧酸酯杂质对照品不仅是对利伐沙班杂质谱的重要补充,而且将对利伐沙班工艺研发中该杂质的监控提供重要参照依据。技术实现要素:发明目的:根据现有的利伐沙班工艺合成路线结合杂质谱研究方法,我们发现利伐沙班噻吩羧酸酯是一种潜在的药物杂质,其可能通过反应过程中噻吩酰氯与中间体醇羟基反应生成,也可能通过利发沙伐药物分子在降解过程中噁唑啉酮五元环发生开环释放出的醇氧根进攻噻吩酰胺键碳基氧同时后发生噻吩酰基迁移造成。按照常规方法可以通过氨基保护,然后再通过羟基酯化反应得到目标物,但实际情况,这种方法脱保护过程中会发生酰基从羟基迁移至氮上。本发明利用噁唑烷在酸性不稳定性,通过控制酸性水解,通过原位氨基质子化抑制氨基的反应活性,快速得到目标杂质。本发明所要解决的技术问题是提供了一种利伐沙班噻吩羧酸酯杂质对照品。该杂质的全称为:1-氨基-3((4-(3-氧代吗啉代)苯基)氨基)丙-2-基5-氯噻吩-2-羧酸乙酯盐酸盐。本发明还要解决的技术问题是提供了一种工艺简单,产品纯度高的制备该杂质对照品的方法,为利伐沙班的质量控制提供合格的利伐沙班噻吩羧酸酯杂质对照品。技术方案:为了解决上述技术问题,本发明提供了一种利伐沙班噻吩羧酸酯杂质对照品,该杂质对照品结构式如式i所示:本发明另一方面提供了上述利伐沙班噻吩羧酸酯杂质对照品的制备方法,该制备方法如下:1)将利伐沙班噁唑烷溶于有机溶剂,然后在酸性水溶液作用下发生开环得到利伐沙班噻吩羧酸酯杂质粗品;2)将利伐沙班噻吩羧酸酯杂质粗品通过有机溶剂重结晶得到高纯度利伐沙班噻吩羧酸酯杂质对照品。本发明通过将利伐沙班噁唑烷溶于有机溶剂然后在酸性条件下开环,制得利伐沙班噻吩羧酸酯粗品,最后通过有机溶剂重结晶得到高纯度的利伐沙班噻吩羧酸酯对照品,具体反应式如下:其中,所述反应有机溶剂为四氢呋喃、丙酮、甲醇、乙醇和乙腈中的一种或几种,优选四氢呋喃;酸性条件是向反应体系中加入盐酸形成的,盐酸的摩尔浓度为1m~5m,优选2m;所述利伐沙班噁唑烷与所述酸的摩尔比为1∶1~1∶5,优化1∶2;反应温度为0℃~50℃,优选25℃;反应时间为3~5小时,优选3小时;重结晶的有机溶剂为甲醇,乙醇和丙醇中的一种或几种,优选甲醇;重结晶温度为25℃~40℃,优选25℃。有益效果:本发明提供的利伐沙班噻吩羧酸酯杂质对照品的制备方法工艺简单,纯度高,能够为利伐沙班的质量控制提供合格的噻吩羧酸酯杂质对照品。利用噁唑烷杂质的噁唑环在酸性条件下容易受到氢质子进攻噁唑氮发生开环,利用开环过程释放出的氨基由于被质子化而抑制酰胺基的形成,可以高效的形成目标杂质的酯键。该合成方法简洁,高效,无需通过额外的官能团保护方法,可以一次性得到高纯度的利伐沙班噻吩羧酸酯杂质对照品。附图说明图1是利伐沙班噻吩羧酸酯杂质对照品的核磁共振氢谱图;图2利伐沙班噻吩羧酸酯杂质质谱图;图3利伐沙班噻吩羧酸酯杂质高效液相谱图。具体实施方式本发明中使用药品来源如下:盐酸和有机溶剂四氢呋喃、丙酮、甲醇、乙醇和乙腈、利伐沙班固体原料由江苏中邦制药有限公司提供;利伐沙班噁唑烷来源于自制,方法如下:1)在500ml反应瓶中依次加入5.0g利伐沙班固体原料和150ml甲醇;2)将2.0g氢氧化钠溶于70ml水中,将制得的氢氧化钠溶液加入到反应瓶中,与反应瓶中的利伐沙班和甲醇混合均匀,在85℃下加热反应5小时,停止反应。用2m盐酸将反应得到的反应液调节至中性,然后用二氯甲烷萃取,萃取的有机相用无水硫酸钠干燥后,旋干有机相,然后将旋干产物真空烘干12h,得到5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺(2.5g)。3)将5-氯-氮-(2-羟基-3-((4-(3-氧杂吗啉)苯基)氨基)丙基)噻吩-2-酰胺2.5g加入250ml单口瓶,然后加入150ml二氯甲烷和2.6g二氯亚砜,25℃搅拌5小时停止反应。将得到的反应液分别用饱和碳酸氢钠溶液、饱和nacl溶液洗涤,然后用无水硫酸钠干燥后旋干,柱层析分离得利伐沙班噁唑烷。对比例1以利伐沙班中间体(i)为起始原料通过常规的保护-脱保护策略来定向合成利伐沙班噻吩羧酸酯杂质(合成路线如下),即首先将(i)里面氨基进行选择性保护(pro代表保护基团,可以是苄基(bn)或则苄氧羰基等)生成中间体(ii),中间体(ii)裸露的羟基与5-氯噻吩酰氯反应生成酯键得到中间体(iii),最后中间体(iii)脱去保护基。但实际结果显示,在脱去保护基的过程中5-氯噻吩酰氯官能团就会发生原位迁移到脱去保护基的氨基上生成化合物(iv)而不是利伐沙班噻吩羧酸酯,主要原因在氨基的反应活性强于羟基,同时生成的酰胺键比酯基更稳定。实施例1利伐沙班噻吩羧酸酯杂质对照品制备方法如下:1)在25ml反应瓶加入0.40g(1mmol)利伐沙班噁唑烷,再加入9ml四氢呋喃,再向反应瓶中加入1ml2m盐酸溶液,25℃搅拌反应4小时,将反应液浓缩干。2)加入8ml甲醇室温溶解,置于冰箱中冷冻过夜,析出白色固体。抽滤,真空干燥,得到噻吩羧酸酯杂质,产率60%。实施例2利伐沙班噻吩羧酸酯杂质对照品制备方法如下:1)在25ml反应瓶加入0.40g(1mmol)利伐沙班噁唑烷,再加入9ml丙酮,再向反应瓶中加入5ml1m盐酸溶液,40℃搅拌反应3小时,将反应液浓缩干。2)加入8ml乙醇室温溶解,置于冰箱中冷冻过夜,析出白色固体。抽滤,真空干燥,得到噻吩羧酸酯杂质,产率56%。实施例3利伐沙班噻吩羧酸酯杂质对照品制备方法如下:1)在25ml反应瓶加入0.40g(1mmol)利伐沙班噁唑烷,再加入9ml乙腈,再向反应瓶中加入1ml3m盐酸溶液,0℃搅拌反应5小时,将反应液浓缩干。2)加入8ml丙醇40℃溶解,置于冰箱中冷冻过夜,析出白色固体。抽滤,真空干燥,得到噻吩羧酸酯杂质,产率52%。实施例4利伐沙班噻吩羧酸酯杂质对照品制备方法如下:1)在25ml反应瓶加入0.40g(1mmol)利伐沙班噁唑烷,再加入9ml甲醇,再向反应瓶中加入1ml3m盐酸溶液,40℃搅拌反应3小时,将反应液浓缩干。2)加入6ml甲醇40℃溶解,置于冰箱中冷冻过夜,析出白色固体。抽滤,真空干燥,得到噻吩羧酸酯杂质,产率58%。实验例:产物表征:(1)对实施例1制备的噻吩羧酸酯杂质对照品进行核磁分析:1h-nmr(500mhz,d6-dmso)8.38(s,3h),7.84(1h,d),7.32(1h,d),7.07(2h,d),6.71(2h,d),6.10(1h,t),5.29(1h,d),4.16(2h,s),3.96(2h,t),3.62(2h,t),3.36-3.52(2h,m),3.29(1h,m),3.19(1h,m)。利伐沙班噻吩羧酸酯杂质对照品的核磁氢谱如图1所示,核磁氢谱上氢质子的数目和化学位移与利伐沙班噻吩羧酸酯杂质结构一致。(2)对实施例1的噻吩羧酸酯杂质对照品进行质谱分析(esi-ms):m/z=410[m+h]+,利伐沙班噻吩羧酸酯杂质质谱如图2所示。(3)对实施例1的噻吩羧酸酯杂质对照品进行高效液相分析利伐沙班噻吩羧酸酯杂质高效液相谱图如图3所示,对应的数据如表1,从表1中可以看出其纯度大于99%。表1检测器a250nm峰号保留时间面积面积%115.69236210.071216.344511315099.929总计5116771100.000通过对核磁质谱谱图进行分析,可以得出结论,所得的终产品为目标产物,且纯度大于99%。本发明对实施例2~4的制备的噻吩羧酸酯杂质对照品进行表征,其结果和实施例1的杂质表征结论一致。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1