优化的联合用药及其治疗癌症和自身免疫疾病的用途的制作方法
本申请是申请日为2016年3月18日、申请号为201680017009.0、发明名称为“优化的联合用药及其治疗癌症和自身免疫疾病的用途”的中国发明专利申请的分案申请。
本发明涉及一系列新的多氟取代的吡唑并嘧啶类化合物及其制备方法,包含它们作为有效成份的药物组合物及其抑制布鲁顿氨酸激酶活性的方法。本发明还涉及多类药物的体外肿瘤细胞活力和体内肿瘤抑制的联合用药优化筛选。本发明的优化联合用药具有协同作用、比单一靶向药物能更好地抑制肿瘤的生存,可造成某些肿瘤完全消失,优化的联合用药比单一靶向药物能更好治疗肿瘤的耐药性和癌症复发。优化的联合用药因剂量低而更安全,并且更好的疗效可造成治疗周期更短。
背景技术:
癌症治疗已经从有毒性化疗发展到联合化疗(如chop),抗体(如利妥昔单抗),以抗体与化疗联合治疗(r-chop),到现在令人兴奋的免疫抗癌疗法(pd-1和pd-l1抗体)和嵌合抗原受体的t细胞疗法(car-t)。患者的寿命和生活质量已有所改善,但由于癌症的复杂性,单一途径或信号通路的突变可以引起一些疗法是无效的或复发。小分子靶向治疗需要每天长期给药,如果停止给药,癌症会反弹或复发。抗体和化疗,给药方式复杂,联合用药可具有累加或协同效应,但很少导致癌症的治愈。靶向治疗与免疫抗癌疗法或嵌合抗原受体的t细胞疗法(car-t)是正在进行中的,但可能具有安全性的担忧。口服疗法有许多优点,比如在遇到严重副作用时可以立即停止给药。抗体或细胞疗法则比较困难马上控制严重副作用。目前单通路靶向治疗或二个靶向通路的联合疗法有一定的局限性,因此癌症的临床疗效(总有效率-orr)大都是部分缓解(pr)或稳定的疾病(误差),很少有治愈(cr)。这种治疗通常会只延长患者生命几个月。此外,细胞疗法的复杂性或抗体化疗的组合也具有安全性问题,以及这些疗法限制于医院环境,不仅导致癌症治疗成本较高,而且影响患者的求治愿望和依从性,及普及到广大患者的局限性。此外,单剂靶向治疗价格每名患者为10-20万美元,新的联合治疗的价格会成倍或更高,这种昂贵的治疗是不可持续发展的。因此,患者和市场都有明确的需要来寻求更好的组合疗法。新的组合疗法或联合用药希望是更为有效与更小的副作用,治疗时间更短。加上合适的伴侣诊断,优化的组合疗法(个性化治疗或精准治疗),可能会治愈癌症亚型,或更有抗耐药性。
ibrutinib(依鲁替尼)是首个被fda批准的布鲁顿酪氨酸激酶(btk)抑制剂,对b细胞淋巴瘤,包括mcl和cll等有良好疗效,但临床治疗已产生c481s等突变而造成耐药性(furmanetal:ibrutinibresistanceinchroniclymphocyticleukemia,nengljmed,2014,2352)。此外,依鲁替尼的药代在患者差异巨大(marosticaetal:populationharmacokineticmodelofibrutinib,abrutontyrosinekinaseinhibitor,inpatientswithbcellmalignancies,cancerchemotherpharmacol(2015)75:111–121),它的毒理动力学研究发现auc在鼠和犬都较低1000h*ng/ml(雄鼠,40mg/kg)和3300h*ng/ml(雌鼠,40mg/kg)及1780h*ng/ml(雄犬,24mg/kg)和1850h*ng/ml(雌犬,24mg/kg)(fda’sndaapplicationnumber205552orig1s000_pharmacologicalreview)。btk起着从细胞表面的b细胞受体(bcr)至下游细胞内反应的b细胞信号传导途径的关键作用。它是b细胞发育,活化,增殖和存活的关键调节剂。
everolimus(依维莫司)是fda批准用于乳腺癌,胰腺癌,肾细胞癌,肾错构瘤和结节性硬化症复合物的治疗mtor激酶抑制剂。mtor蛋白的机制作用已被完全阐明,它是在一个非常复杂的信号传导途径中控制细胞生长,增殖,代谢和细胞凋亡的关键调节剂。此外器官移植也会激活mtor,依维莫司在低剂量下用于治疗器官移植的排斥。
pomalidomide(泊马度胺)是在2013年fda批准治疗多发性骨髓瘤的免疫调节药物。泊马度胺和有关同类imids抑制细胞因子tnf-α,il-1β,il-6,il-12和gm-csf。除了它们的抗血管生成,抗增殖和促凋亡性质外,它们也刺激t淋巴细胞诱导其增殖,t细胞因子的产生,和t细胞毒性,从而增加了t细胞的抗癌活性。
venetoclax或abt-199是处于临床研究中的bcl-2抑制剂。b淋巴细胞瘤-2(bcl-2)蛋白对细胞凋亡有关键作用。abt-199临床中曾引发肿瘤溶解综合症(tls)而导致患者死亡。
methotrexate(甲氨蝶呤)为抗叶酸类抗肿瘤药和治疗类风湿关节炎药物,主要通过对二氢叶酸还原酶的抑制而达到阻碍细胞的合成,而抑制细胞的生长与繁殖。
布鲁顿氨酸激酶(bruton’styrosinekinase,btk)属于tec家族的成员。它由独特的n-端结构域即ph(pleckstrinhomology)结构域、th(techomology)同源区、sh3(srchomology3)结构域、sh2(srchomology2)结构域和催化结构域,也称sh1/tk(srchomology1/tyrosinekinase)结构域或者激酶结构域组成(akinleyeetal:ibrutinibandnovelbtkinhibitorsinclinicaldevelopment,journalofhematology&oncology2013,6:59)。在b淋巴细胞正常发育过程中,btk基因不同蛋白区域的正确表达在b细胞的功能及多种转导途径中具有关键性作用。
btk功能下游有多种受体,包括生长因子,b细胞抗原,趋化因子和先天免疫受体,从而启动一个多元化范围内的细胞过程,如细胞增殖,存活,分化,运动,血管生成,细胞因子生产,抗原表达等。因此btk在许多造血细胞信号传输途径中起重要作用,同时在b细胞激活,发育,存活和信号传导中也至关重要(kurosaki,molecularmechanismsinbcellantigenreceptorsignaling.curropimm,1997,9(3):309-18)。
已有证据证明b细胞对自身免疫应答和炎性反应有免疫调节作用。如cd20抗体利妥昔单抗(rituxan)是消耗b细胞的基于蛋白质的治疗剂,用作治疗自身免疫性疾病如慢性淋巴细胞白血病和自身抗体导致的炎性疾病如类风湿关节炎。因此通过抑制b细胞活化过程中发挥关键作用的蛋白质激酶应该可以对于b细胞相关疾病病理有帮助。
btk在自身免疫疾病中的作用的证据已经由btk-缺失型小鼠和btk-充足型小鼠模型试验提供(killp,etal:bruton’styrosinekinasemediatedsignalingenhancesleukemogenesisinamousemodelforchroniclymphocyticleukemia.amjbloodres2013,3(1):71–83.)。在慢性淋巴细胞白血病(cll)小鼠模型中,btk-缺失型小鼠完全废止慢性淋巴细胞白血病,btk过度表达会加速白血病发病,增加死亡率。
已知btk抑制剂的选择性不理想:除了抑制btk,还抑制其他多种激酶(如etk,egf,blk,fgr,hck,yes,brk和jak3等),从而可能产生较多的副作用。选择性更好的抑制剂副作用可能更小。
已知btk抑制剂体内生成多种衍生物,这也会影响药效和副作用。已知btk抑制剂的药代动力学也可改善。
技术实现要素:
本发明涉及btk抑制剂用作治疗或抑制自身免疫性疾病或病症、异种免疫性疾病或病症、炎性疾病以及癌症或病症的方法。包括给所述患者施用有效剂量由式(i)、(ii)、(ia)、(iia)、(ib)和(iib)表达的化合物,或它们的药学上可接受的盐。
其中:
r1为氟原子;n为1,2,3或4;
r2为氟原子;m为1或2;
r3为氢原子或氘原子。
本发明中,术语“药学上可接受的盐”指的是以酸或碱形式存在的盐,其非受限的实例为(a)酸性加成盐,无机酸(例如,盐酸、氢溴酸、硫酸、磷酸、硝酸等),有机酸的盐,有机酸例如醋酸、草酸(oxalicacid)、酒石酸(tartaricacid)、琥珀酸(succinicacid)、苹果酸(malicacid)、抗坏血酸、苯甲酸(benzoicacid)、鞣酸(tannicacid)、扑酸(pamoicacid)、海藻酸(alginicacid)、聚麸胺酸(polyglutamicacid)及水杨酸等;(b)碱性加成盐(baseadditionsalts),形成有金属阳离子,如锌、钙、钠、钾等。
合成方法
本发明通过实施例和本文中所公开的化合物来示例。在本发明的具体化合物选自下组被公开的实施例中的化合物和它们的药学上可接受的盐及它们的单独的非对映体的化合物或盐。
本发明对合成路径进行了积极探索,成功设计了合成吡唑并嘧啶类化合物的新方法(见方案1-2和具体反应实例)。
除非另有说明,在下面的反应方案和讨论中,r1,r2,r3,m,n的定义如上所述。
反应式方案1
在碱的存在下,如碳酸钾,并在适当的溶剂中,如dmf,氟取代的起始材料a1可以与取代的苯酚b1生成中间体c1。在适当的碱,如醋酸钾的存在下,并在适当的溶剂,如1,4-二氧六环中,中间体c1可与双联频哪醇硼酸酯在适当的催化剂(如[1,1'-双(二苯基磷)二茂铁]二氯化钯)作用下进行反应以提供中间d1。在适当的溶剂中,如dmf,起始原料1h-吡唑并[3,4-d]嘧啶-4-胺可与nis进行反应得到中间体f1,然后经由mitsunobu反应或者取代反应形成中间体g1。在适当的碱,如磷酸钾的存在下,并在适当的溶剂,如1,4-二氧六环和水中,中间体g1可与硼酸酯d1在适当的催化剂(如pd-118)作用下进行反应得到中间h1。在酸性条件下脱去h1的boc基团,得到胺化合物i1。胺化合物i1可以与亲电子试剂反应,得到产物i。当i为外消旋体时可通过sfc手性拆分得到具有光学活性的化合物ia和ib。
反应式方案2
在碱的存在下,如碳酸钾,并在适当的溶剂中,如dmf,3-氟-4-溴苯酚与1-氟-3硝基苯生成中间体c2。在适当的还原剂,如铁粉和氯化铵,并在适当的溶剂中,如乙醇和水,硝基化合物c2可被还原成胺d2,随后在亚硝酸钠和吡啶氟化氢的作用下可生成氟取代的中间体e2。在适当的碱,如醋酸钾的存在下,并在适当的溶剂,如1,4-二氧六环中,e2可与双联频哪醇硼酸酯在适当的催化剂(如[1,1'-双(二苯基磷)二茂铁]二氯化钯)作用下进行反应以提供硼酸酯f2。在适当的碱,如磷酸钾的存在下,并在适当的溶剂,如1,4-二氧六环和水中,中间体h1可与硼酸酯f2在适当的催化剂(如pd-118)作用下进行反应以提供中间g2。在酸性条件下脱去g2的boc基团得到胺化合物h2。胺化合物h2可以与亲电子试剂反应,得到产物i2。当i2为外消旋体时可通过sfc手性拆分得到光学活性的化合物j2和k2。
反应式方案3
3-氟-4-溴苯酚与3-氟-苯硼酸在适当的催化剂(如醋酸铜)作用下生成中间体b3,随后b3可与双联频哪醇硼酸酯在适当的催化剂(如[1,1'-双(二苯基磷)二茂铁]二氯化钯)作用下进行反应以提供硼酸酯c3。在适当的碱,如醋酸钾的存在下,并在适当的溶剂,如1,4-二氧六环和水中,中间体g1可与硼酸酯c3在适当的催化剂(如[1,1'-双(二苯基磷)二茂铁]二氯化钯)作用下进行反应以提供中间d3。在酸性条件下脱去d3的boc基团得到胺化合物e3。胺化合物e3可以与亲电子试剂反应,得到产物f3。当f3为外消旋体时可通过sfc手性拆分得到光学活性的化合物h3和i3。
反应式方案4
胺化合物i1可以与丁-2炔酸反应,得到产物ii。当ii为外消旋体时可通过sfc手性拆分得到具有光学活性的化合物iia和iib。
反应式方案5
中间体f1经由mitsunobu反应或者取代反应形成有光学活性的中间体a5。在适当的碱,如磷酸钾的存在下,并在适当的溶剂,如1,4-二氧六环和水中,中间体a5可与硼酸酯d1在适当的催化剂(如pd-118)作用下进行反应得到中间b5。在酸性条件下脱去b5的boc基团,得到胺化合物c5。胺化合物c5可以与亲电子试剂反应,得到产物ia。
反应式方案6
胺化合物c5可以与丁-2炔酸反应,得到产物iia。
本发明提供了由通式(i)、(ii)、(ia)、(iia)、(ib)和(iib)所述化合物,及它们的对映体和非对映体或其可药用的盐。
本发明由通式(i)、(ii)、(ia)、(iia)、(ib)和(iib)所述化合物包括含有一个或者多个稳定同位素或放射同位素,所述同位素包括,但不仅限于2h,3h,13c,14c,15n,18o等。
本发明首次将1h的同位素2h引入btk抑制剂。
本发明由通式(i)、(ii)、(ia)、(iia)、(ib)和(iib)所述的化合物乙烯基的双键末端1h可被2h替代,从而降低由双键氧化还原而导致的药物失活。
本发明提供了制备由通式(i)、(ii)、(ia)、(iia)、(ib)和(iib)所述化合物,及它们的对映体和非对映体的方法。
本发明提供了一种用于调节btk的活性,并且治疗或抑制与btk活性相关的疾病的方法。经证实,本发明的化合物对btk活性具有抑制作用,本发明提供了由通式(i)、(ii)、(ia)、(iia)、(ib)和(iib)所述化合物用于治疗和/或预防以下疾病的药剂中的活性成分,所述的疾病为由不利的细胞因子信号传导引发的疾病,这些疾病包括,但不限于:
(1)自身免疫性疾病(autoimmunediseases)如慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasturesyndrome)、寻常天皰疮、类天皰疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等,系统性红斑狼疮、类风湿关节炎、银屑病(psoriasis)、系统性脉管炎、硬皮病、天疱疮、混合结缔组织病、自身免疫性溶血性贫血、甲状腺自身免疫病、溃疡性结肠炎等;
(2)异常免疫性疾病如血清病、哮喘、过敏性鼻炎和药物过敏等;
(3)炎性疾病如角膜炎、鼻炎、口腔炎、腮腺炎、咽炎、扁桃体炎、气管炎、支气管炎、肺炎、心肌炎、胃炎、肠胃炎、胆囊炎、阑尾炎等;
(4)癌症包括,但不限于各种b细胞恶性肿瘤(包括小淋巴细胞淋巴瘤(sll)、慢性淋巴细胞白血病(cll)、弥漫性大b细胞淋巴瘤(dlbcl)、华氏巨球蛋白血症(waldenstrommacroglobulinemia)、滤泡性淋巴瘤(follicularlymphom)、套细胞淋巴瘤(mcl))以及其他抑制btk激酶活性对病人有益处的疾病;
其他抑制btk激酶活性对病人有益处的疾病包括但不限于:脑瘤,膀胱癌,胃癌,卵巢癌,胰腺癌,乳腺癌,头颈癌,子宫颈癌,子宫内膜癌,直肠癌,肾癌,食道癌,前例腺癌,甲状腺癌,骨癌,皮肤癌,结肠癌,雌性生殖道瘤,淋巴瘤,多发性骨髓瘤(mm)和睾丸癌等。
所述方法包括给需要的患者有效剂量的如权利要求1-2中的化合物。
可以根据标准的药学实践,将本发明的化合物(btk抑制剂)的药物制剂单独使用或者与一种或多种另外的药剂联合使用,所述btk抑制剂的药物制剂与所述另外的药剂的给药途径可以相同或不同,并且给药时间可以相同或不同。所述的另外的药剂包括(但不限于)酪氨酸激酶抑制剂(如阿西替尼、达沙替尼、埃克替尼等)、拓扑异构酶抑制剂(如托泊替康等)、蛋白激酶c抑制剂(如aeb-071等)、鞘氨醇-1-磷酸受体激动剂(如芬戈莫德、krp-203等)、抗t细胞免疫球蛋白(如atgam等)、抗il-2受体的抗体(如达利珠单抗等)、酰胺(ctx)、异环磷酰胺(ifo)、阿霉素(adm)、柔红霉素(dnr)、长春新碱(vcr)、长春花碱(vbl)、依托泊甙(vp16)、威猛(vumon)、卡铂(cbp)和甲氨蝶呤(mtx)环孢菌素a、他克莫司、西罗莫司、依维莫司、硫唑嘌呤、布喹那、来氟米特、lea-29y、抗cd3抗体(如0kt3等)、阿司匹林、b7-cd28阻断分子(如belatacept、abatacept等)、cd40-cd154阻断分子(例如抗cd40抗体等)、扑热息痛、布洛芬、萘普生、吡罗昔康和抗炎甾体类(例如氢化泼尼松或地塞米松)。
布鲁顿的酪氨酸激酶(btk)抑制剂包括所有已知的、但不仅限于:依鲁替尼ibrutinib,化合物3和5,acp-196(acertapharma),bgb-3111(beigene),avl-292(celgene),ono-4059(onepharmaceutical),hm71224(hanmipharmaceutical),rn486(roche),cnx-774和cgi-11746等。
mtor抑制剂包括所有已知的、但不仅限于:依维莫司,雷帕霉素,xl388,gdc-0349,azd2014,azd8055,gsk105965,mln0128和ridaforlimus等。
免疫调节药物imids包括所有已知的、但不仅限于:thalidomide(沙利度胺),revlimid(来那度胺),pomalidomide(泊马度胺),cc-122,cc-220。
pi3k激酶抑制剂包括所有已知的、但不仅限于:btg226,pf-05212384(gedatolisib),gdc-0980(apitolisib),gsk2126458,bez235,ipi-145(duvelisib)和cal-101(idelalisib)等。
bcl-2抑制剂包括所有已知的、但不仅限于:abt-199(venetoclax)和bi-97c1(sabutoclax)等。
topk抑制剂包括所有已知的、但不仅限于:ots964(oncotherapyscience)。
jak3激酶抑制剂包括所有已知的、但不仅限于:托法替尼(tofactinib)。
jak1/2激酶抑制剂包括所有已知的、但不仅限于:鲁索替尼(ruxolitinib)。
alk激酶抑制剂包括所有已知的、但不仅限于:克唑替尼(crizotinib),色瑞替尼(ceritinib)和ch5424802(alectinib)。
本发明也涉及具体的多种组合药物在体外细胞和动物体内肿瘤模型优化筛选。体外能杀死肿瘤细胞的化合物或化合物组合,因为药代药谢问题,并不一定在动物肿瘤模型中有效。因此在没有动物体内肿瘤抑制前提下、仅有体外细胞数据不能证明化合物或化合物组合能否成药。本发明采取全为口服灌胃的给药方式(而非腹腔注射或静脉注射)而解决了药代药谢不确定因素,对化合物或化合物组合在多种动物肿瘤模型进行筛选优化而达到成药性的评估。
现有技术声称alk抑制剂和btk抑制剂有协同抑制体外肿瘤细胞效应,但本发明显示了相反结果:alk抑制剂色瑞替尼(ceritinib)和btk抑制剂依鲁替尼ibrutinib没有协同效应。
现有技术也声称jak2抑制剂和btk抑制剂有协同抑制体外肿瘤细胞效应,但本发明显示了相反结果:jak1/2抑制剂鲁索替尼(ruxolitinib)不但和btk抑制剂、mtor抑制剂、免疫调节药物imid对肿瘤细胞都没有协同效应,对tmd-8肿瘤细胞基本上无抑制作用。
本发明公开了优越于单一靶向药物的二合一和三合一的联合用药(复方用药或组合药物)。二合一和三合一的联合用药在更低剂量具有协同效应。本发明涉及的联合用药配方比例不仅限于本发明公开的体外和体内实验的具体比例。
本发明同时对控制细胞生长,增殖,存活和凋亡的多个关键信号通路靶向进行优化组合筛选。单一靶向治疗经常因基因突变而产生耐药性造成药无效并导致疾病复发。联合用药可以克服耐药性而达到更好的治疗效果或治愈。本发明显示联合用药对肿瘤细胞有协同效应和合成致死能力,其的体外效力比单一靶向药物的活性高达100倍的增加。在多种移植瘤模型抑制口服灌胃给药,联合用药还展示了更卓越的疗效,在比每个单一靶向药物剂量低得多的组合给药条件下(例如:btk抑制剂3的十八分之一剂量,mtor抑制剂依维莫司的六分之一剂量,imid免疫调节剂泊马度胺的六分之一剂量),联合用药的疗效远远优于每个单一靶向药物的疗效。二合一的联合用药在15天的治疗周期可以导致肿瘤完全消退,而三合一的联合用药在更短的治疗周期(9天)可以导致肿瘤完全消退,并且在停止给药后12天内没有观察肿瘤反弹,疗效显著优于比单一靶向治疗。单一靶向治疗不但需要长期治疗,而且常有肿瘤的反弹和耐药性的产生以及癌症复发。联合用药因为使用比单一靶向药物低得多的剂量并同时展现更卓越的疗效将使优化的联合用药比每个单药都拥有更好的安全性和更广泛的治疗窗。本发明联合用药的上述独特性能可能为难以治疗的癌症患者带来新希望。
本发明三合一药物组合在btk抑制剂低到10nm浓度下可高达95%肿瘤细胞活力抑制是意想不到的,不是现有发明可推测或指导的。细胞活力抑制是在温育48小时后测试,如果温育时间增长到72-96小时,组合药物将显示更强的细胞活力抑制。本发明显示体外细胞活力抑制与体内肿瘤抑制相对应,体外细胞活力抑制可以预测体内肿瘤抑制效果。测试中最低浓度下(如10nm)的高效应体外细胞活力抑制可导致肿瘤消褪或者完全消失。低至10nm浓度下有很高肿瘤细胞活力抑制可杀死肿瘤细胞的发明具有巨大的临床治疗应用。在治疗患者疗程中,人体的肿瘤细胞内组合药物很容易到达10-100nm浓度,从而得到更好的治疗效果。目前单一靶向治疗或双向联合治疗通常在化合物浓度达到1000nm左右才能有效地抑制肿瘤细胞的生长。比如abt-199在1000nm、100nm和10nm浓度下抑制tmd-8细胞活力分别为37.6%、18.8%和11.1%。在和btk抑制剂3的二合一对tmd-8细胞活力抑制分别为85.97%、79.99%和65.36%。而在和btk抑制剂3及pi3k抑制剂的三合一对tmd-8细胞活力高效抑制分别为95.56%、95.30%和94.62%。和btk抑制剂3及mtor抑制剂依维莫司的三合一对tmd-8细胞活力也有高效抑制,分别为93.44%、94.73%和94.65%。本发明的超强协同效应不是现有技术可推测或指导的。
本发明提供了(1)布鲁顿的酪氨酸激酶(btk)抑制剂和mtor激酶抑制剂及imid免疫调节药物的三合一药物组合为联合用药最佳组合之一;另外两个最佳组合的三合一药物组合分别为(2)鲁顿的酪氨酸激酶(btk)抑制剂和mtor激酶抑制剂及bcl-2抑制剂;(3)鲁顿的酪氨酸激酶(btk)抑制剂和pi3k抑制剂及bcl-2抑制剂。其次分别为布鲁顿的酪氨酸激酶(btk)抑制剂和mtor激酶抑制剂的二合一药物组合、布鲁顿的酪氨酸激酶(btk)抑制剂和免疫调节药物imid的二合一药物组合、布鲁顿的酪氨酸激酶(btk)抑制剂和topk抑制剂的二合一药物组合、topk抑制剂和pi3k激酶抑制剂的二合一药物组合、布鲁顿的酪氨酸激酶(btk)抑制剂和pi3k激酶抑制剂的二合一药物组合。
本发明显示了某些组合抑制有协同效应,某些组合没有协同效应和起相反作用。细胞分子信号通道过多并交叉而复杂,同时抑制多信号通道的组合更多更复杂。因此本发明所述三种最佳三合一组合的超强协同效应不是现有技术可推测或指导的.
本发明提供了体外细胞活力抑制和动物体内肿瘤抑制及多种肿瘤模型的结合优化筛选方法,解决了组合化合物成药性评价的难题。
本发明显示,在口服灌胃动物肿瘤模型中只有三合一的药物组合[布鲁顿的酪氨酸激酶(btk)抑制剂、mtor激酶抑制剂和imid免疫调节药物]和二合一的药物组合[布鲁顿的酪氨酸激酶(btk)抑制剂和mtor激酶抑制剂]的联合用药可以导致肿瘤完全消失,而且在停止给药后没有观察到肿瘤反弹生长。其他组合药物只抑制肿瘤增长,必须持续给药才能抑制肿瘤增长。虽然单药依维莫司在中高剂量也可导致肿瘤完全消失,但在停止给药后肿瘤快速反弹生长,这是靶向治疗的特征和缺陷。本发明解决了该缺陷,有望个体化治愈癌症。
本发明不仅在敏感的tmd-8肿瘤模型上验证了肿瘤抑制的卓越疗效,也在不敏感的dohh2肿瘤模型和耐药而难治的wsu-dlcl肿瘤模型分别证明了三合一组合药物的良好疗效。虽然在该二种模型上没有导致肿瘤完全消失,三合一组合药物的疗效也是最优秀的。比如epizyme公司论文显示在wsu-dlcl肿瘤模型上,化合物ez-6438(组蛋白甲基转移酶histonemethyltransferaseezh2抑制剂)需每天给小鼠灌胃非常高剂量(每公斤480毫克)才能达到类似疗效。而本发明三合一组合药物用药仅需每天给小鼠灌胃每公斤21毫克。
本发明第一次证明多靶向联合用药具有更好的疗效,并大大降低了单一药物的剂量,因此可以进一步降低每个单药的副作用,而使联合用药更安全更有效。
本发明的多靶向联合用药不仅可用于b-细胞引起的恶性淋巴肿瘤和血癌等,也可治疗t-细胞引起的癌症及固体肿瘤等。
本发明的多靶向联合用药也可用于治疗类风湿关节炎等自身免疫疾病。
本发明还提供了含有布鲁顿的酪氨酸激酶(btk)抑制剂、mtor激酶抑制剂和imid免疫调节剂的三者作为活性成分的药物组合物。
本发明还提供了含有布鲁顿的酪氨酸激酶(btk)抑制剂、mtor激酶抑制剂和bcl-2抑制剂的三者作为活性成分的药物组合物。
本发明还提供了含有布鲁顿的酪氨酸激酶(btk)抑制剂、pi3k激酶抑制剂和bcl-2抑制剂的三者作为活性成分的药物组合物。
本发明还提供了将式(i)、(ii)、(ia)、(iia)、(ib)和(iib)表示的化合物用于制备调节btk活性、治疗或者抑制与btk活性相关疾病的药物的用途。
本发明还提供了将式(i)、(ii)、(ia)、(iia)、(ib)和(iib)表示的化合物与mtor激酶抑制剂和/或imid免疫调节药物联合用于制备调节btk活性、治疗或者抑制与btk活性相关疾病的药物的用途。
本发明还提供了将布鲁顿的酪氨酸激酶(btk)抑制剂和mtor激酶抑制剂和imid免疫调节药物的三者组合、或者其中两者组合,用于制备调节btk活性、治疗或者抑制与btk活性相关疾病的药物的用途。
本发明还提供了将布鲁顿的酪氨酸激酶(btk)抑制剂和mtor激酶抑制剂和bcl-2抑制剂的三者组合,用于制备调节btk活性或bcl-2活性、治疗或者抑制与btk或bcl-2活性相关疾病的药物的用途。
本发明也提供了将布鲁顿的酪氨酸激酶(btk)抑制剂和pi3k激酶抑制剂和bcl-2抑制剂的三者组合,用于制备调节btk活性或pi3k活性或bcl-2活性、治疗或者抑制与btk活性或pi3k活性或bcl-2活性相关疾病的药物的用途。
可使用载体、赋形剂和其它通常用于药物制剂的添加剂,来制备含有一种或两种或多种式(i)、(ii)、(ia)、(iia)、(ib)和(iib)所示的化合物或其可药用盐作为活性成分的药物组合物。
可采用片剂、丸剂、胶囊、颗粒剂、散剂、乳剂、糖浆、混悬剂或液体制剂等的口服给药的剂型,或者采用静脉注射或肌肉注射、栓剂、皮下剂、透皮剂、经鼻剂、吸入剂等的非经口给药的剂型,来进行治疗给药。应考虑要治疗的各个患者的症状、年龄、性别等以便适当地确定化合物的剂量。通常来说,在口服给药的情况下,成人患者每日服用的化合物剂量为0.001mg/kg至100mg/kg左右,并将该剂量一次服用或分为2~4次服用。在根据症状需要采用静脉给药的情况中,通常来说,成人患者按照每次0.0001mg/kg至10mg/kg的剂量范围,每日一次至多次给药。另外,在采用吸入剂给药的情况中,通常来说,成人患者按照每次0.0001mg/kg至lmg/kg的剂量范围,每日一次至多次给药。
在本发明中,用于经口给药的固体组合物可以采用片剂、散剂、颗粒剂等剂型。在这种固体组合物中,将一种或一种以上的活性物质与至少一种惰性赋形剂(例如乳糖、甘露糖醇、葡萄糖、羟丙基纤维素、微晶纤维素、淀粉、聚乙烯吡咯烷酮、硅酸镁铝等)混合。按照常规方法,组合物中也可以含有惰性添加剂,如润滑剂(如硬脂酸镁)、崩解剂(如羧甲基淀粉钠)和溶解助剂。根据需要,片剂或丸剂也可以被糖衣或者胃溶性或肠溶性包衣剂所包覆。
用于经口给药的液体组合物包括可药用的乳液剂、溶液制剂、混悬剂、糖浆剂、或酏剂等,并含有通常可使用的惰性稀释剂(例如纯化水、乙醇)。除所述惰性稀释剂以外,该组合物中也可以含有诸如增溶剂、润湿剂、悬浮剂之类的助剂以及甜味剂、矫味剂、芳香剂、防腐剂。
用于非经口给药的注射剂包括无菌的水性或非水性溶液制剂、混悬剂、乳剂。用于水性溶液的稀释剂可(例如)包括注射用蒸馏水和生理盐水。用于非水性溶液的稀释剂可(例如)包括丙二醇、聚乙二醇、植物油(如橄榄油)、醇类(如乙醇)和聚山梨醇酯80。这样的组合物中还可以含有诸如等渗剂、防腐剂、润湿剂、乳化剂、分散剂、稳定剂、溶解助剂之类的添加剂。可以采用通过可截留细菌的过滤器进行过滤、添加杀菌剂或进行光辐射的方法来对所述组合物进行灭菌。此外,也可以将这些组合物制成无菌的固体组合物,在使用前再用无菌水或无菌的注射用溶剂来使其溶解或混悬而使用。
诸如吸入剂和经鼻剂之类的经粘膜剂,可以以固体、液体、或半固体的状态使用,并且可以按照以往公知的方法来制备这些经粘膜剂。例如,可以根据需要而添加赋形剂(如乳糖和淀粉)、ph调节剂、防腐齐ij、表面活性剂、润滑齐ij、稳定剂和增稠剂等。给药时可以使用合适的吸入或吹送用装置。例如,可以使用计量给药吸入装置等公知的装置或喷雾器,将化合物单独地或作为配方后的混合物粉末来给药。此外,也可以将化合物与可药用的载体组合后,作为溶液或混悬液来给药。干燥粉末吸入器等可以用于单次给药或多次给药,并可以使用干燥粉末或含有粉末的胶囊。另外,也可以采用加压气溶胶喷雾等形式,通过使用适当的抛射剂(例如,氯氟烷烃、氢氟烷烃、或二氧化碳等适当的气体)来给药。
表1.btk抑制剂的化合物
注:如果在画出的结构和给出的该结构的名称之间有差异,则以画出的结构为准。
附图说明
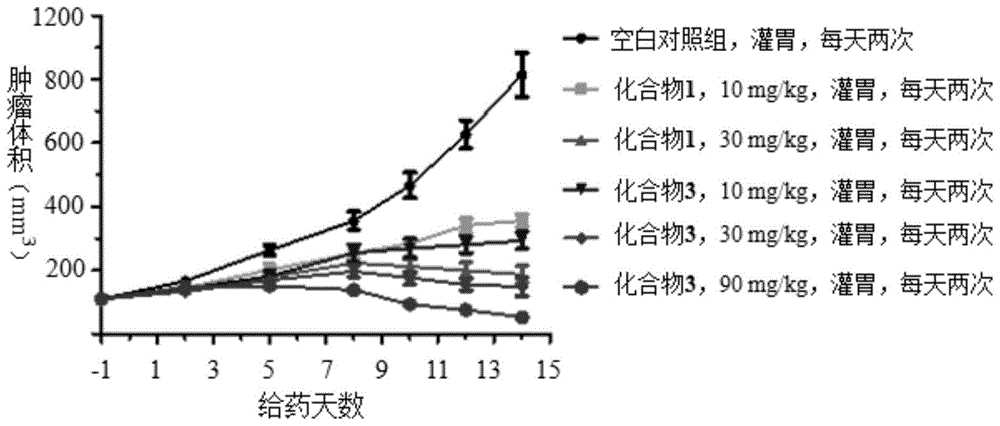
图1-a.化合物1和3多剂量在scid小鼠体内tmd-8模型中的肿瘤体积抑制作用,其中给药方式:灌胃,每天两次;给药天数:14天。
图1-b.化合物1和3多剂量在scid小鼠体内tmd-8模型中的肿瘤重量抑制作用,其中给药方式:灌胃,每天两次;给药天数:14天。
图2.化合物3和15及联合用药在scid小鼠体内tmd-8肿瘤模型中的抗肿瘤作用。
图3.化合物3、8、15及联合用药在scid小鼠体内tmd-8肿瘤模型中的抗肿瘤作用。
图4.化合物3、14、16及联合用药在scid小鼠体内tmd-8肿瘤模型中的抗肿瘤作用,其中
图5.化合物3、14、15及联合用药在scid小鼠体内tmd-8肿瘤模型中的抗肿瘤作用,其中
图6.化合物3、14、15及联合用药在scid小鼠体内dohh-2肿瘤模型中的抗肿瘤作用。
图7.化合物3、14、15及联合用药在scid小鼠体内dohh-2肿瘤模型中的抗肿瘤作用。
图8.化合物3和9在耐药性的wsu-dlcl2肿瘤模型中的抗肿瘤作用。
图9.化合物3、14、15及联合用药在scid小鼠体内耐药性的wsu-dlcl2肿瘤模型中的抗肿瘤作用。
图10.化合物3、14和15的“三合一”联合用药在小鼠体内tmd-8肿瘤模型中的抗肿瘤作用。
图11.化合物3、14、15与化合物9、14、15的“三合一”联合用药在小鼠体内dohh2肿瘤模型中的抗肿瘤作用。
图12.化合物3、8、12及“二合一”或“三合一”联合用药在小鼠体内tmd-8肿瘤模型中的抗肿瘤作用。
图13.足体积--佐剂诱发关节炎。
图14.病理组织切片--佐剂诱发关节炎。
图15.临床评分--胶原诱导性关节炎。
图16.病理组织切片--胶原诱导性关节炎。
发明详述
实施例1
化合物1和2
1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮
1-[(s)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮
步骤a:
3-(4-溴-3-氟苯氧基)-1,2,4,5-四氟苯
操作步骤:
向化合物3-氟-4-溴苯酚(47.0g,246.1mmol,1.0eq.)的dmf(500ml)溶液中加入碳酸钾(68.0g,492.1mmol,2.0eq.)和化合物1,2,3,4,5-五氟苯(49.6g,295.3mmol,1.2eq.)。反应液在100℃搅拌12小时,减压蒸除溶剂。将残余物溶于乙酸乙酯(300ml)中,用水(100ml)洗两次,饱和食盐水(100ml)洗一次。有机相用无水硫酸钠干燥,浓缩旋干得到目标化合物(78g,收率:93%)。
步骤b:
2-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷
操作步骤:
将化合物3-(4-溴-3-氟苯氧基)-1,2,4,5-四氟苯(73g,215.3mmol,1.0eq.),双联频哪醇硼酸酯(65.6g,258.4mmol,1.2eq.),醋酸钾(31.6g,322.9mmol,1.5eq.)和[1,1'-双(二苯基磷)二茂铁]二氯化钯(9.4g,12.8mmol,0.06eq.)溶于1,4-二氧六环(1l)中,反应液在80℃,并在氮气保护下搅拌14小时。冷却至室温后,反应液用硅藻土过滤,滤液旋干,得到的粗品用硅胶柱层析纯化分离(展开剂:石油醚)得到目标化合物(60g,收率:72%)。
步骤c:
3-碘-1h-吡唑并[3,4-d]嘧啶-4-胺
操作步骤:
向1h-吡唑并[3,4-d]嘧啶-4-胺(100g,0.74mol,1.0eq.)的dmf(800ml)溶液中加入nis(250g,1.11mol,1.5eq.)。反应液在氮气保护下在80~85℃搅拌16小时。反应液过滤,滤饼用乙醇洗涤三次,每次1000ml,得到目标化合物(184g,收率:95%)。
步骤d:
3-(甲基磺酰氧基)哌啶-1-甲酸叔丁酯
操作步骤:
在0℃下向3-羟基哌啶-1-甲酸叔丁酯(10.0g,50mmol,1.0eq.)的二氯甲烷(100ml)溶液中依次滴加三乙胺(15g,150mmol,3.0eq.)和甲基磺酰氯(6.3g,55mmol,1.1eq.)。反应液在20℃搅拌1小时,加入饱和nahco3(100ml),然后用二氯甲烷(200ml)萃取三次。有机相用无水硫酸钠干燥,浓缩旋干得到目标化合物(13g,收率:95%)。
步骤e:
3-(4-氨基-3-碘-1h-吡唑并[3,4-d]-嘧啶-1-基)哌啶-1-甲酸叔丁酯
操作步骤:
在0℃下向3-碘-1h-吡唑并[3,4-d]-嘧啶-4-胺(8.1g,31mmol,1.0eq.)的dmf(50ml)溶液中加入碳酸铯(20.2g,62mmol,2.0eq.)和3-(甲基磺酰氧基)哌啶-1-甲酸叔丁酯(13g,46.5mmol,1.5eq.)。反应液在80℃搅拌过夜,用硅藻土过滤,浓缩旋干,得到的粗品用硅胶柱层析纯化分离(洗脱剂:乙酸乙酯)得到目标化合物(5g,收率:25%)。
步骤f:
3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-甲酸叔丁酯
操作步骤:
将化合物3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)哌啶-1-甲酸叔丁酯(7.6g,17.1mmol,1.0eq.),2-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷(8.6g,22.3mmol,1.3eq.),磷酸钾(7.3g,34.2mmol,2.0eq.)和pd-118(0.56g,0.855mmol,0.05eq.)溶于1,4-二氧六环/水(240ml,5:1,v/v)中。反应在氮气保护下,在60℃反应12小时。冷却至室温后,将反应液倾入冰水(300ml)中,然后用乙酸乙酯(100ml)萃取四次。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品用硅胶柱层析纯化分离(展开剂:乙酸乙酯)得到目标化合物(6.8g,收率:69%)。
步骤g:
3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺
操作步骤:
在0℃下向3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-甲酸叔丁酯(6.8g,11.8mmol)的乙酸乙酯(50ml)溶液中加入hcl/ea(20ml,4mol/l)。反应液在室温下搅拌1小时,浓缩旋干得到目标化合物的盐酸盐(5.2g,收率:86%)。
步骤h:
1-[3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮
操作步骤:
在0℃下向3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺(1.5g,2.9mmol,1.0eq.)的二氯甲烷(10ml)溶液中依次滴加三乙胺(887mg,8.7mmol,3.0eq.)和丙烯酰氯(0.26g,2.9mmol,1.0eq.)。反应液在0℃搅拌1小时,用水(5ml)淬灭,然后用二氯甲烷(50ml)稀释,水(30ml)洗两次,饱和食盐水(30ml)洗一次。有机相用无水硫酸钠干燥,浓缩旋干,所得粗品用硅胶柱层析分离纯化(洗脱剂:石油醚:乙酸乙酯=1:0~1:1)得到目标化合物(0.94g,收率:64%)。
光谱数据:
lc/ms(方法:uflc):rt=3.130分钟;m/z=531.1[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,dmso-d6)δ8.22(s,1h),8.00-7.91(m,1h),7.55-7.46(m,1h),7.27(dd,j=2.4,10.8hz,1h),7.12(dd,j=2.4,8.8hz,1h),6.88-6.65(m,1h),6.13-6.02(m,1h),5.70-5.56(m,1h),4.71-4.65(m,1h),4.54-4.51(m,0.5h),4.20-4.17(m,1h),4.07-4.04(m,0.5h),3.67-3.60(m,0.5h),3.17-3.12(m,1h),2.98-2.94(m,0.5h),2.26-2.21(m,1h),2.11-2.06(m,1h),1.92-1.89(m,1h),1.58-1.54(m,1h).
步骤i:
1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮
1-[(s)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮
操作步骤:
将化合物1-[3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮(750mg)进行手性sfc分离(chiralceloj,20μm;supercriticalco2:c2h5oh(0.2%dea),v/v,200ml/min)得到目标化合物1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮(280mg,ee:100%)和1-[(s)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-三氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮(330mg,ee:98%)。
光谱数据:
化合物1:
lc/ms(方法:uflc):rt=3.002分钟;m/z=531.1[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cdcl3)δ8.36(s,1h),7.58(t,j=8.4hz,1h),7.09-7.04(m,1h),6.94-6.88(m,2h),6.62-6.54(m,1h),6.32-6.25(m,1h),5.73-5.63(m,1h),5.56-5.51(m,1h),4.90-4.85(m,1.5h),4.59-4.56(m,0.5h),4.21-4.17(m,0.5h),4.04-4.01(m,0.5h),3.76-3.71(m,0.5h),3.40-3.35(m,0.5h),3.22-3.15(m,0.5h),2.93-2.87(m,0.5h),2.39-2.27(m,2h),2.04-1.68(m,2h).
化合物2:
lc/ms(方法:uflc):rt=3.006分钟;m/z=531.1[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cd3od)δ8.24(s,1h),7.62(t,j=8.4hz,1h),7.50-7.45(m,1h),7.09-7.01(m,2h),6.85-6.63(m,1h),6.21-6.09(m,1h),5.77-5.61(m,1h),4.63-4.59(m,1h),4.23-4.07(m,1.5h),3.90-3.85(m,0.5h),3.51-3.45(m,0.5h),3.34-3.17(m,1.5h),2.40-2.23(m,2h),2.08-2.05(m,1h),1.75-1.71(m,1h).
实施例2
化合物3
1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
方法一:
步骤a:
(r)-3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)吡咯烷-1-甲酸叔丁酯
操作步骤:
在0℃并在氮气保护下,向化合物3-碘-1h-吡唑并[3,4-d]嘧啶-4-胺(24g,92mmol,1.0eq.),化合物(s)-3-羟基-吡咯烷-1-甲酸叔丁酯(26g,137.5mmol,1.5eq)和pph3(36g,137.5mmol,1.5eq.)的四氢呋喃(720ml)溶液中滴加diad(27.6g,137.5mmol,1.5eq.)。反应液在0℃搅拌1小时,然后在室温下搅拌过夜。减压蒸除溶剂,向反应瓶中加入乙腈(200ml),然后室温下搅拌2小时,过滤,滤饼用乙腈(20ml)洗涤,干燥得到目标化合物(25g,收率:63%)。
步骤b:
(3r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-甲酸叔丁酯
操作步骤:
将化合物(r)-3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)吡咯烷-1-甲酸叔丁酯(25g,58mmol,1.0eq.),化合物2-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷(30g,75.4mmol,1.3eq.),磷酸钾(25g,116mmol,2.0eq.)和pd-118(750mg,1.16mmol,0.02eq.)溶于1,4-二氧六环/水(600ml,5:1,v/v)中。反应液在氮气保护下,在60℃搅拌过夜。冷却至室温后,用硅藻土过滤。滤液浓缩旋干,向剩余物中加入水(300ml),然后用乙酸乙酯萃取三次,每次300ml。合并的有机相用无水硫酸钠干燥,浓缩旋干得到目标化合物(60g,粗品)。
步骤c:
3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺
操作步骤:
在0℃下向(3r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-甲酸叔丁酯(60g,粗品)的乙酸乙酯(100ml)溶液中加入hcl/ea(100ml,4mol/l)。反应液在室温下搅拌1小时,浓缩旋干得到目标化合物的盐酸盐。向反应瓶中加入水(500ml),用乙酸乙酯萃取三次,每次300ml。水相调节ph=9,用乙酸乙酯萃取三次,每次300ml。合并的有机相用无水硫酸钠干燥,浓缩旋干得到目标化合物(24g,两步收率:90%)。
步骤d:
1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
操作步骤:
在-5℃下向3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺(23.5g,50.75mmol,1.0eq.)的四氢呋喃(470ml)溶液中加入naoh(10%,94ml),然后滴加丙烯酰氯(5.97g,66mmol,1.3eq.)。反应液在-5℃搅拌1小时,用饱和食盐水(100ml)淬灭,用乙酸乙酯萃取三次,每次200ml。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品用硅胶柱层析分离纯化(洗脱剂:石油醚:乙酸乙酯=1:3~1:1),得到产品溶于甲醇(500ml),过滤。向搅拌的滤液中加水(1500ml),然后搅拌2小时,过滤,滤饼减压干燥得到目标化合物3(16.5g,收率:63%)。
光谱数据:
lc/ms(方法:uflc):rt=3.764分钟;m/z=517.0[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cd3od)δ8.45(s,1h),7.70(t,j=8.4hz,1h),7.55-7.46(m,1h),7.12-7.05(m,2h),6.70-6.55(m,1h),6.33-6.26(m,1h),5.81-5.75(m,1h),4.23-3.83(m,5h),2.68-2.55(m,2h).
方法二:
操作步骤:
在0℃下向3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺(1.0g,2.16mmol,1.0eq.)的四氢呋喃(50ml)和水(10ml)溶液中加入naoh(216mg,5.40mmol,2.5eq.),然后滴加氯丙酰氯(288mg,2.27mmol,1.05eq.)的四氢呋喃溶液(10ml)。反应液在0℃搅拌1小时,然后升温至60℃搅拌12小时。冷却至室温后加饱和食盐水(10ml),用乙酸乙酯萃取三次,每次50ml。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品用硅胶柱层析分离纯化(洗脱剂:石油醚:乙酸乙酯=1:3~1:1)得到目标化合物3(0.8g,收率:71%)。
方法三:
操作步骤:
将化合物(r)-[3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)-1-吡咯烷基]-2-丙烯-1-酮(100g,0.26mmol,1.0eq.),化合物2-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷(120mg,0.31mmol,1.2eq.),碳酸钠(55mg,0.52mmol,2.0eq.)和pd(pph3)4(30mg,0.026mmol,0.01eq.)溶于1,4-二氧六环/水(5ml,1:1,v/v)中。反应液在微波照射下在80℃搅拌30分钟。冷却至室温后,用硅藻土过滤。滤液浓缩旋干,得到的粗品采用高效液相色谱在c18反相柱上分离(流动相:乙腈/水/0.5%hcl,梯度洗脱10%至100%(体积比)),减压蒸除易挥发的组分后冻干得到目标化合物(38mg,产率:28%)。
方法四:
化合物3和化合物4
1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
1-[(s)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
步骤a:
3-(甲基磺酰氧基)吡咯烷-1-甲酸叔丁酯
操作步骤:
在0℃下向3-羟基吡咯烷-1-甲酸叔丁酯(30.0g,163mmol,1.0eq.)的二氯甲烷(200ml)溶液中依次滴加三乙胺(35g,346mmol,2.1eq.)和甲基磺酰氯(36.6g,321mmol,1.9eq.)。反应液在0℃搅拌3小时,用水(20ml)淬灭,然后用水(100ml)洗两次,饱和食盐水(100ml)洗一次。有机相用无水硫酸钠干燥,浓缩旋干得到目标化合物(45.6g,收率:100%)。
步骤b:
3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)吡咯烷-1-甲酸叔丁酯
操作步骤:
向化合物3-(甲基磺酰氧基)吡咯烷-1-甲酸叔丁酯(35g,134mmol,3.5eq.)的dmf(300ml)溶液中加入碳酸铯(37g,115mmol,3.0eq.)和化合物3-碘-1h-吡唑并[3,4-d]嘧啶-4-胺(10g,38mmol,1.0eq.)。反应液在85℃搅拌12小时,冷却至室温后过滤。滤液浓缩旋干,所得粗品用硅胶柱层析纯化分离(洗脱剂:石油醚:乙酸乙酯=1:1)得到目标化合物(7.0g,收率:44%)。
步骤c:
3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-甲酸叔丁酯
操作步骤:
将化合物3-(4-胺-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)吡咯烷-1-甲酸叔丁酯(8g,18mmol,1.0eq.),2-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷(10.7g,27mmol,1.5eq.),磷酸钾(7.6g,36mmol,2.0eq.)和pd-118(1.2g,1.8mmol,0.1eq.)溶于1,4-二氧六环/水(180ml,5:1,v/v)中。反应在氮气保护下,在60℃搅拌14小时。冷却至室温后,将反应液倾入冰水(50ml)中,用乙酸乙酯(100ml)萃取三次。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品用硅胶柱层析分离纯化(洗脱剂:乙酸乙酯:石油醚=1:1)得到目标化合物(2.5g,收率:25%)。
步骤d:
3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-(吡咯烷-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺
操作步骤:
在0℃下向3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-甲酸叔丁酯(2.5g,4.4mmol)的二氯甲烷(20ml)溶液中加入hcl/ea(20ml,4mol/l)。反应液在室温下搅拌1小时,浓缩旋干得到目标化合物的盐酸盐(2.2g,收率:100%)。
步骤e:
1-[3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
操作步骤:
在0℃下向3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-(吡咯烷-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺(2.2g,4.4mmol,1.0eq.)的二氯甲烷(50ml)溶液中依次滴加三乙胺(1.4g,12.8mmol,3.0eq.)和丙烯酰氯(0.38g,4.2mmol,0.95eq.)。反应液在0℃搅拌1小时,用水(30ml)淬灭,分液,水相用二氯甲烷(30ml)萃取三次。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品用硅胶柱层析纯化分离(洗脱剂:乙酸乙酯)得到目标化合物(1.0g,收率:45%)。
光谱数据:
lc/ms(方法:uflc):rt=2.810分钟;m/z=517.1[m+h]+;总的运行时间为7分钟。
步骤f:
1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
1-[(s)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
操作步骤:
化合物1-[3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-基]丙-2-烯-1-酮通过sfc手性拆分得到化合物3(270mg)和化合物4(320mg)。
光谱数据:
lc/ms(方法:uflc):rt=2.808分钟;m/z=517.1[m+h]+;总的运行时间为7分钟。
实施例3
化合物5
1-[(r)-3-[4-氨基-3-[2-氟-4-(3-氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
步骤a:
1-(3-氟-4-溴苯氧基)-3-硝基苯
操作步骤:
向化合物3-氟-4-溴苯酚(40g,210mmol,1.0eq.)的dmf(400ml)溶液中加入1-氟-3硝基苯(29.6g,210mmol,1.0eq.)和碳酸钾(58g,420mmol,2.0eq.)。反应液在氮气保护下并在90℃搅拌12小时,减压蒸除溶剂。向剩余物中加水(300ml)然后用乙酸乙酯(300ml)萃取三次。合并的有机相用无水硫酸钠干燥,浓缩旋干得到目标化合物(65g,收率:100%)。
步骤b:
3-(3-氟-4-溴苯氧基)苯胺
操作步骤:
向化合物1-(3-氟-4-溴苯氧基)-3-硝基苯(65g,210mmol,1.0eq.)的乙醇(300ml)和水(60ml)溶液中加入氯化铵(28g,525mmol,2.5eq.)和铁粉(58.8g,1.05mol,5.0eq.)。反应液在氮气保护下回流搅拌12小时,冷却至室温后用硅藻土过滤,滤液浓缩旋干,得到的粗品采用高效液相色谱在c18反相柱上分离(流动相:乙腈/水/0.7%nh4hco3,梯度洗脱10%至100%(体积比)),减压蒸除易挥发的组分后冻干得到目标化合物(19g,产率:23%)。
步骤c:
1-溴-2-氟-4-(3-氟苯氧基)苯
操作步骤:
在-10℃向吡啶-氟化氢溶液(30ml)中分批加入化合物3-(3-氟-4-溴苯氧基)苯胺(9g,32mmol,1.0eq.)。所得反应液在0℃搅拌30分钟,然后冷却在-10℃分批加入亚硝酸钠(2.42g,35mmol,1.1eq.)。反应液在20℃搅拌30分钟,然后在60℃搅拌14小时。冷却至室温后,将反应液倾入冰-乙醇(50ml)中,加入nahco3饱和溶液(50ml),然后用乙酸乙酯(50ml)萃取三次,合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品用硅胶柱层析分离纯化(洗脱剂:石油醚)得到目标化合物(5.8g,产率:64%)。
步骤d:
2-[2-氟-4-(3-氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷
操作步骤:
将化合物1-溴-2-氟-4-(3-氟苯氧基)苯(5.8g,20mmol,1.0eq.),双联频哪醇硼酸酯(6.1g,24mmol,1.2eq.),醋酸钾(3.9g,40mmol,2.0eq.)和[1,1'-双(二苯基磷)二茂铁]二氯化钯(0.89g,1.2mmol,0.06eq.)溶于1,4-二氧六环(100ml)中,反应液在85℃,并在氮气保护下搅拌14小时。冷却至室温后,反应液用硅藻土过滤,滤液旋干,得到的粗品用硅胶柱层析纯化分离(洗脱剂:石油醚)得到目标化合物(6.5g,收率:100%)。
步骤e:
(3r)-3-[4-氨基-3-[2-氟-4-(3-氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-甲酸叔丁酯
操作步骤:
将化合物3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)吡咯烷-1-甲酸叔丁酯(6.5g,15.0mmol,1.0eq.),化合物2-[2-氟-4-(3-氟苯氧基)苯基]-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷(6.5g,19.6mmol,1.3eq.),磷酸钾(6.4g,30.1mmol,2.0eq.)和pd-118(0.25g,0.39mmol,0.01eq.)溶于1,4-二氧六环/水(16ml,1:1,v/v)中。反应液在氮气保护下,在85℃搅拌12小时。冷却至室温后,反应液用水(50ml)稀释,然后用乙酸乙酯(100ml)萃取三次。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品采用硅胶柱层析分离纯化(洗脱剂:乙酸乙酯)得到目标化合物(4.2g,产率:55%)。
步骤f:
3-[2-氟-4-(3-氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺
操作步骤:
在0℃下向(3r)-3-[4-氨基-3-[2-氟-4-(3-氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]吡咯烷-1-甲酸叔丁酯(4.2g,8.27mmol)的二氯甲烷(15ml)溶液中加入hcl/ea(10ml,4mol/l)。反应液在室温下搅拌1小时,浓缩旋干得到目标化合物的盐酸盐(3.7g,收率:92%)。
步骤g:
1-[(r)-3-[4-氨基-3-[2-氟-4-(3-氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-2-丙烯-1-酮
操作步骤:
在0℃下向3-[2-氟-4-(3-氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺(3.7g,8.27mmol,1.0eq.)的四氢呋喃(20ml)溶液中依次滴加氢氧化钠溶液(10%,15.3ml)和丙烯酰氯(0.67g,7.44mmol,0.9eq.)。反应液在室温下搅拌10分钟,用饱和nahco3(20ml)淬灭,用二氯甲烷(30ml)萃取三次。合并的有机相用无水硫酸钠干燥,浓缩旋干,得到的粗品采用硅胶柱层析分离纯化(洗脱剂:石油醚:乙酸乙酯=1:0~1:1)得到目标化合物(2.5g,产率:65%)。
光谱数据:
lc/ms(方法:uflc):rt=3.178分钟;m/z=463.0[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cdcl3)δ8.36(s,1h),7.53-7.49(m,1h),7.40-7.35(m,1h),6.95-6.81(m,4h),6.41-6.39(m,2h),5.69-5.55(m,3h),4.14-3.98(m,3h),3.78-3.72(m,1h),2.71-2.54(m,2h).
实施例4
化合物6
(e)-1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-3-氘-2-丙烯-1-酮
步骤a:
(e)-3-溴丙烯酸
操作步骤:
化合物丙炔酸(1g,14.28mmol,1.0eq.)和hbr(40%水溶液,1.7ml,0.88eq.)的混合液在140℃搅拌过夜。减压蒸除溶剂,所得粗品用水结晶三次,每次4ml,得到目标化合物(0.76g,产率:35%)。
光谱数据:
1hnmr(400mhz,cdcl3)δ7.76(d,j=14hz,1h),6.55(d,j=14hz,1h).
步骤b:
(e)-3-氘丙烯酸
操作步骤:
在0~5℃下向化合物(e)-3-溴丙烯酸(3g,19.87mmol,1.0eq.)的d2o(30ml)中加入na-hg(6g,49.67mmol,2.5eq.)。反应液在室温下搅拌36小时。分液,水相用1m的盐酸调ph=5,然后用乙醚萃取五次,每次20ml。合并的有机相用无水硫酸钠干燥,减压蒸除溶剂得到目标化合物(0.52g,产率:36%)。
光谱数据:
1hnmr(400mhz,cdcl3)δ7.76(d,j=17.2hz,1h),6.55(d,j=17.2hz,1h).
步骤c:
(e)-1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-3-氘-2-丙烯-1-酮
操作步骤:
向化合物3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺(500mg,1.08mmol,1.0eq.)的二氯甲烷(50ml)溶液中加入(e)-3-氘丙烯酸(76mg,1.08mmol,1.0eq.),hatu(530mg,1.40mmol,1.3eq.)和n,n-二异丙基乙胺(419mg,3.24mmol,3.0eq.)。反应液在室温下搅拌12小时,浓缩旋干,得到的粗品采用高效液相色谱在c18反相柱上分离(仪器:lc8a&gilson215,组分收集柱:synergimax-rp150*30mm*4u,流动相a:水(0.5%hcl),流动相b:乙腈,流速:30ml/min,梯度:b36%至37%,0-17分钟),减压蒸除易挥发的组分后冻干得到目标化合物盐酸盐(76mg,产率:13%)。
光谱数据:
lc/ms(方法:uflc):rt=2.765分钟;m/z=518.1[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cd3od)δ8.41(s,1h),7.66(t,j=8.4hz,1h),7.51-7.44(m,1h),7.09-7.01(m,2h),6.66-6.56(m,1h),6.28-6.23(m,1h),5.75-5.66(m,1h),4.19-4.16(m,1h),4.06-4.02(m,1.5h),3.89-3.85(m,1h),3.78-3.72(m,0.5h),2.63-2.49(m,2h).
实施例5
化合物7
(z)-1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-3-氘-2-丙烯-1-酮
步骤a:
(z)-3-溴丙烯酸
操作步骤:
化合物丙炔酸(1g,14.28mmol,1.0eq.)和hbr(40%水溶液,1.7ml,0.88eq.)的混合液在55℃搅拌过夜。减压蒸除溶剂,所得粗品用石油醚结晶三次,每次4ml,得到目标化合物(0.3g,产率:14%)。
光谱数据:
1hnmr(400mhz,cdcl3)δ7.16(d,j=8.4hz,1h),6.67(d,j=8.4hz,1h).
步骤b:
(z)-3-氘丙烯酸
操作步骤:
在0~5℃下向化合物(z)-3-溴丙烯酸(3g,19.87mmol,1.0eq.)的d2o(30ml)中加入na-hg(6g,49.67mmol,2.5eq.)。反应液在室温下搅拌36小时。分液,水相用1m的盐酸调ph=5,然后用乙醚萃取五次,每次20ml。合并的有机相用无水硫酸钠干燥,减压蒸除溶剂得到目标化合物(0.34g,产率:23%)。
光谱数据:
1hnmr(400mhz,cdcl3)δ6.14(d,j=10.4hz,1h),5.96(d,j=10.4hz,1h).
步骤c:
(z)-1-[(r)-3-[4-氨基-3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]-3-氘-2-丙烯-1-酮
操作步骤:
向化合物3-[2-氟-4-(2,3,5,6-四氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺(1.0g,2.16mmol,1.0eq.)的二氯甲烷(50ml)溶液中加入(z)-3-氘丙烯酸(151mg,2.16mmol,1.0eq.),hatu(1.06g,2.80mmol,1.3eq.)和n,n-二异丙基乙胺(838mg,6.48mmol,3.0eq.)。反应液在室温下搅拌12小时,浓缩旋干,得到的粗品采用高效液相色谱在c18反相柱上分离(流动相:乙腈/水/0.5%hcl,梯度洗脱36%至37%(体积比)),减压蒸除易挥发的组分后冻干得到目标化合物盐酸盐(228mg,产率:20%)。
光谱数据:
lc/ms(方法:uflc):rt=2.775分钟;m/z=518.1[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cd3od)δ8.45(s,1h),7.70(t,j=8.4hz,1h),7.52-7.46(m,1h),7.13-7.05(m,2h),6.71-6.61(m,1h),5.80-5.73(m,2h),4.23-4.20(m,1h),4.09-4.04(m,1.5h),3.93-3.90(m,1h),3.80-3.75(m,0.5h),2.67-2.56(m,2h).
实施例6
化合物7x
1-[(r)-3-[4-氨基-3-[2-氟-4-(3-氟苯氧基)苯基]-1h-吡唑并[3,4-d]嘧啶-1-基]-1-吡咯烷基]丁-2-炔-1-酮
操作步骤:
向3-[2-氟-4-(3-氟苯氧基)苯基]-1-[(r)-吡咯烷-3-基]-1h-吡唑并[3,4-d]嘧啶-4-胺(200.00mg,489.72umol,1.00eq.)的二氯甲烷(5.0ml)溶液中依次加入丁-2炔酸(41.17mg,489.72umol,1.00eq.)hatu(93.10mg,244.86umol,0.50eq.)和dipea(75.95mg,587.66umol,102.64ul,1.20eq.)。反应液在15-18℃搅拌2小时,浓缩旋干。得到的粗品采用高效液相色谱在c18反相柱上分离(流动相:乙腈/水/0.5%hcl,梯度洗脱22%至52%(体积比)),减压蒸除易挥发的组分后冻干得到目标化合物盐酸盐(82mg,产率:33%)。
光谱数据:
lc/ms(方法:uflc):rt=3.057分钟;m/z=475.0[m+h]+;总的运行时间为7分钟。
1hnmr(400mhz,cdcl3)δ9.92(s,1h),8.34(d,j=8.8hz,1h),7.56(br,1h),7.41-7.36(m,1h),7.00-6.86(m,5h),6.58(br,1h),5.62-5.58(m,1h),4.22-3.74(m,4h),2.65-2.50(m,2h),2.02-1.96(m,3h).
生物体外实验部分
btk激酶活性抑制实验:
btk野生型激酶标准htrf实验的酶反应液组成为1nmbtk野生型激酶,1μm生物素-tk1多肽,30μmatp和50mmhepes(ph值7.5)缓冲液。混合物在室温下孵育60分钟后,加入edta将反应停止。然后将抑制剂(5l)中加入终浓度为2nm抗体和62.5nm的xl665。将板在室温下孵育60分钟后使用envision多标记微孔板检测仪读取实验结果。检测数据通过公式(minratio)/(max-min)*100%转化为抑制百分率(%),随后通过四个参数曲线拟合得到测试化合物的ic50值。
体外肿瘤细胞活力抑制实验:
肿瘤细胞(tmd-8,dohh2和wsu-dlcl2)转移并附着到96孔板。过夜后,加入空白对照缓冲液和选择浓度(0.01nm-100μm)的测试化合物溶液。在48小时温育后,再加入celltiter-go试剂裂解细胞。记录发光信号并计算细胞活力的抑制百分比。
动物实验方法
鼠药代实验:雄性sd大鼠24小时内的药物代谢动力学试验共分为静脉和口服两组,每组3只动物.静脉组取血时间点为给药前,给药后0.0833,0.167,0.5,1,2,4,8,24小时;口服组取血时间点为给药前,给药后0.167,0.5,1,2,4,8,24小时。采血完毕后,利用hplc-ms/ms进行生物分析,报告化合物的血药浓度.计算出的药物代谢动力学参数为静脉组动物的平均清除率(clp),平均表观分布容积(vdss),0-24h曲线下分布面积(auc),0-24小时的平均滞留时间(mrt),半衰期t1/2;口服组动物在给药后的平均最大药物浓度(cmax),0-24h曲线下分布面积(auc),0-24小时的平均滞留时间(mrt);本次试验的平均相对生物利用度。
犬药代实验:比格犬24小时内的药物代谢动力学试验共分为静脉(每公斤1毫克)和口服(每公斤3毫克)两组,每组3只动物.静脉组取血时间点为给药前,给药后0.033,0.083,0.25,0.5,1,3,6,9,24小时;口服组取血时间点为给药前,给药后0.083,0.25,0.5,1,3,6,9,24小时。采血完毕后,利用hplc-ms/ms进行生物分析,报告化合物的血药浓度.计算出的药物代谢动力学参数为静脉组动物的平均清除率(clp),平均表观分布容积(vdss),0-24h曲线下分布面积(auc),0-24小时的平均滞留时间(mrt),半衰期t1/2;口服组动物在给药后的平均最高药物浓度(cmax),0-24h曲线下分布面积(auc),0-24小时的平均滞留时间(mrt);平均相对生物利用度。
单药及联合用药在动物体内tmd-8,dohh2或wsu-dlcl2肿瘤模型中的抑制作用:
实验结果显示,联合给药具有协同药效作用,对敏感的(tmd-8)、难治性的(dohh-2)以及多耐药性的(wsu-dlcl2)肿瘤模型均表现出优于单一药物的抑制效果。
采用雌性cb-17scid小鼠异种移植模型评估化合物(3,9,14及其他表中列出的化合物)及其联合用药的抑瘤效果。将tmd-8,dohh2,wsu-dlcl2肿瘤细胞接种于含有10%热灭活胎牛血清的rpmi-1640培养基中,在37℃,5%co2条件下培养。肿瘤细胞常规传代培养,每周两次。当细胞生长进入指数生长期时收获并计数,以进行肿瘤接种。每只小鼠右侧皮下接种0.2ml肿瘤细胞pbs悬液(10x106)和基质胶(1:1)。当平均肿瘤体积大约为100-200mm3时开始给药。每组由6-10只荷瘤小鼠组成。测试组(包括空白对照组、单药组、联合给药组)按照预定剂量口服给药,连续给药14天或者21天。实验过程中,小鼠体重和肿瘤体积每两天或三天测量一次。
胶原诱导性关节炎模型:
采用雄性dba/1小鼠胶原诱导性关节炎模型评估化合物3和化合物14联合用药的体内抑制效果。小鼠分为8组,包括一个正常组、一个溶剂对照组、5个治疗组。所有小鼠(除正常组)在第0天和21天用200μg牛胶原(ii型)免疫。加强免疫后七天(28天),动物开始表现出症状的疾病,平均临床评分约1。同日,免疫小鼠随机分为7组:化合物3(1.5mpk)和化合物14(0.15mpk)联合给药,一天两次;化合物3(4.5mpk)和化合物14(0.45mpk)联合给药,一天两次;化合物3(1.5mpk)和化合物14(0.15mpk)联合给药,一天一次;化合物3(1.5mpk)单药,一天一次;化合物14(1.5mpk)单药,一天一次;阳性对照组(0.2mg/kg地塞米松),并开始给药。口服给药两周,记录体重和临床评分。在研究结束时,处死动物,收集后肢进行组织病理学分析。
佐剂性关节炎模型:
采用雌性lewis大鼠胶原诱导性关节炎模型评估化合物3和化合物14联合用药的体内抑制效果。所有大鼠(除正常组)在第0天左后肢注射完全弗氏佐剂(cfa)进行免疫。在免疫后6天,一些大鼠开始显示关节炎的临床症状,如红斑和肿胀。在第13天,免疫的动物被重新组合为7组:溶剂对照组,化合物3(5mpk)和化合物14(0.5mpk)联合给药,一天两次;化合物3(15mpk)和化合物14(1.5mpk)联合给药,一天两次;化合物3(30mpk)和化合物14(3mpk)联合给药,一天一次;化合物3(5mpk)单药,一天两次;化合物14(0.5mpk)单药,一天两次;阳性对照组(化合物11,3mpk,一天两次),并开始给药。口服给药三周,每隔一天记录体重、爪体积和临床评分。在研究结束时,处死动物,收集右后爪,采用h.e.染色进行组织病理学分析。
表2.本发明实施例化合物抑制btk酶的活性数据表
表3.单一化合物对tmd-8细胞株抑制百分比(inh%)
注:本表中的化合物3、6、7、8-19对应于相应的“化合物中文名或英文名”中所指明的化合物,其中3、6、7指代本发明实施例中的化合物,8-19指代现有技术中相应的化合物。为了便于行文,下文以编号表示相应的化合物,因此,以下化合物编号具有相同的含义。
表4.二合一组合化合物对tmd-8细胞株抑制百分比(inh%)
由表4可以看出,“二合一”联合用药具有显著肿瘤细胞活力抑制作用。其中,化合物8+化合物13,化合物14+化合物13,化合物15+化合物13以及化合物3+化合物13的药物组合均呈现了对tmd-8细胞活力的高效抑制作用。
表5.三合一组合化合物对tmd-8细胞株抑制百分比(inh%)
由表5可以看出,“三合一”联合用药具有显著肿瘤细胞活力抑制作用。其中,化合物3+化合物14+化合物12和化合物3+化合物8+化合物12的药物组合在btk抑制剂低至10nm浓度下仍具有高达95%肿瘤细胞活力抑制作用。
表6.单一化合物和三合一组合化合物对耐药性的wsu-dlcl2细胞株抑制百分比(inh%)
由表6可以看出,化合物3+化合物15+化合物14的“三合一”药物组合对多耐药性的wsu-dlcl2细胞具有良好细胞活力抑制作用,并且明显优于每个单一靶向药物。
表7.单一化合物和三合一组合化合物对dohh-2细胞株抑制百分比(inh%)
由表7可以看出,化合物3+化合物15+化合物14的“三合一”药物组合对难治性的dohh-2细胞具有良好细胞活力抑制作用,并且明显优于每个单一靶向药物。
表8.不同比例组合化合物对tmd-8细胞株抑制百分比(inh%)
由表8可以看出,不同比例的“三合一”药物组合对敏感的tmd-8细胞均具有显著细胞活力抑制作用
表9.化合物3在大鼠体内的pk参数
表10.化合物3在犬体内的pk参数
表11.化合物3在大鼠体内的tk参数
表12.化合物3在犬体内的tk参数
表13.单药及联合用药在动物体内肿瘤模型中的抑瘤作用
由表13可以看出,联合用药对肿瘤细胞有协同效应和合成致死能力,在比每个单一靶向药物剂量低得多的组合给药条件下,联合用药的疗效远远优于每个单一靶向药物的疗效。例如,化合物3+化合物14“二合一”的联合用药在15天的治疗周期可以导致肿瘤完全消退,而化合物3+化合物14+化合物15“三合一”的联合用药在更短的治疗周期(9天)可以导致肿瘤完全消退,并且在停止给药后12天内没有观察肿瘤反弹,疗效显著优于比单一靶向治疗。
表14.联合用药在动物体内肿瘤模型中体重影响
由表14可以看出,各组动物体重无差异,低剂量联合用药安全。
表15.联合用药3/14/15在动物体内肿瘤模型中体重影响
由表15可以看出,各组动物体重无差异,低剂量联合用药安全。
表16.足体积--佐剂诱发关节炎
*p<0.05,**p<0.01,***p<0.001
由表16可以看出,“二合一”联合用药比单药更有效。
表17.病理评分--佐剂诱发关节炎
***p<0.001,v.s.空白对照组,kruskal-wallistest,dunn’spost-hoctest
由表17可以看出,“二合一”联合用药比单药更有效。
表18.初次免疫后第21天之后的平均临床评分--胶原诱导性关节炎
由表18可以看出,“二合一”联合用药比单药更有效。
表19.病理评分--胶原诱导性关节炎
***p<0.001,v.s.空白对照组,kruskal-wallistest,dunn’spost-hoctest
由表19可以看出,“二合一”联合用药比单药更有效。
本发明还包括下述具体实施方案:
1.由通式(i)、(ii)、(ia)、(iia)、(ib)和(iib)表示的化合物,它们的对映体和非对映体或其可药用的盐
其中:
r1为氟原子;n为1,2,3或4;
r2为氟原子;m为1或2;
r3为氢原子或氘原子。
2.由下列结构式表示的化合物,它们的消旋物或其可药用的盐
3.一种药物组合物,它包括一种惰性载体和具体实施方案1-2中任一项所述的化合物或其可药用的盐,优选具体实施方案2的化合物。
4.一种药物,它包括一种惰性载体和具体实施方案1-2中任意一项所述的化合物或其可药用的盐作为活性成分,优选具体实施方案2的化合物。
5.一种抑制在患者体内btk活性的方法,包括给予所述患者有效剂量的如具体实施方案1-2中的化合物。
6.一种用于治疗或抑制自身免疫性疾病(autoimmunediseases)或病症的方法,所述方法包括给需要的患者有效剂量的如具体实施方案1-2中的化合物或具体实施方案3的组合物,所述自身免疫性疾病包括,但不限于器官特异性自身免疫疾病,如慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasturesyndrome)、寻常天皰疮、类天皰疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等;系统性自身免疫疾病,如系统性红斑狼疮、类风湿关节炎、银屑病(psoriasis)、系统性脉管炎、硬皮病、天疱疮、混合结缔组织病、自身免疫性溶血性贫血、甲状腺自身免疫病、溃疡性结肠炎等。
7.一种用于治疗或抑制异常免疫性疾病或病症的方法,所述方法包括给需要的患者有效剂量的如具体实施方案1-2中的化合物或具体实施方案3的组合物,所述异常免疫性疾病包括,但不限于血清病、哮喘、过敏性鼻炎和药物过敏等。
8.一种用于治疗或抑制炎性疾病或病症的方法,所述方法包括给需要的患者有效剂量的如具体实施方案1-2中的化合物或具体实施方案3的组合物,所述炎性疾病包括,但不限于角膜炎、鼻炎、口腔炎、腮腺炎、咽炎、扁桃体炎、气管炎、支气管炎、肺炎、心肌炎、胃炎、肠胃炎、胆囊炎、阑尾炎等。
9.一种用于治疗或抑制癌症或其他疾病的方法,所述方法包括给需要的患者有效剂量的如具体实施方案1-2中的化合物或具体实施方案3的组合物,所述癌症或其他疾病包括,但不限于各种b细胞恶性肿瘤(包括小淋巴细胞淋巴瘤(sll)、慢性淋巴细胞白血病(cll)、弥漫性大b细胞淋巴瘤(dlbcl)、华氏巨球蛋白血症(waldenstrommacroglobulinemia)、滤泡性淋巴瘤(follicularlymphom)、套细胞淋巴瘤(mcl)和多发性骨髓瘤(mm))以及其他抑制btk激酶活性对病人有益处的疾病。
10.具体实施方案1-2的化合物在制备治疗或抑制自身免疫性疾病(autoimmunediseases)的药物中的用途,所述自身免疫性疾病包括,但不限于器官特异性自身免疫疾病,如慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasturesyndrome)、寻常天皰疮、类天皰疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等,系统性自身免疫疾病,如系统性红斑狼疮、类风湿关节炎、银屑病(psoriasis)、系统性脉管炎、硬皮病、天疱疮、混合结缔组织病、自身免疫性溶血性贫血、甲状腺自身免疫疾病、溃疡性结肠炎等。
11.具体实施方案1-2的化合物在制备治疗或抑制异常免疫性疾病的药物中的用途,所述异常免疫性疾病包括,但不限于血清病、哮喘、过敏性鼻炎和药物过敏等。
12.具体实施方案1-2的化合物在制备治疗或抑制炎性疾病的药物中的用途,所述炎性疾病包括,但不限于角膜炎、鼻炎、口腔炎、腮腺炎、咽炎、扁桃体炎、气管炎、支气管炎、肺炎、心肌炎、胃炎、肠胃炎、胆囊炎、阑尾炎等。
13.具体实施方案1-2的化合物在制备治疗癌症或其他疾病的药物中的用途,其中所述癌症或其他疾病包括,但不限于各种b细胞恶性肿瘤(包括小淋巴细胞淋巴瘤(sll)、慢性淋巴细胞白血病(cll)、弥漫性大b细胞淋巴瘤(dlbcl)、华氏巨球蛋白血症(waldenstrommacroglobulinemia)、滤泡性淋巴瘤(follicularlymphom)、套细胞淋巴瘤(mcl)和多发性骨髓瘤(mm))以及其他抑制btk激酶活性对病人有益处的疾病。
14.具体实施方案1-2的化合物和各种cd20抗体联合用药在制备治疗癌症或其他疾病的药物中的用途,其中所述癌症或其他疾病包括,但不限于各种b细胞恶性肿瘤(包括小淋巴细胞淋巴瘤(sll)、慢性淋巴细胞白血病(cll)、弥漫性大b细胞淋巴瘤(dlbcl)、华氏巨球蛋白血症(waldenstrommacroglobulinemia)、滤泡性淋巴瘤(follicularlymphoma)、、套细胞淋巴瘤(mcl)和多发性骨髓瘤(mm))以及其他抑制btk激酶活性对病人有益处的疾病。
15.一种三合一的药物组合物,其中含有btk抑制剂、mtor抑制剂以及imid免疫调节剂。
16.具体实施方案15所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、mtor抑制剂everolimus(依维莫司)以及imid免疫调节剂thalidomide(沙利度胺)/revlimid(来那度胺)/pomalidomide(泊马度胺)/cc-122/cc-220。
17.具体实施方案15所述的三合一药物组合物为具体实施方案2中所述的化合物3、mtor抑制剂everolimus(依维莫司)以及imid免疫调节剂thalidomide(沙利度胺)/revlimid(来那度胺)/pomalidomide(泊马度胺)/cc-122/cc-220。
18.具体实施方案15所述的三合一药物组合物为具体实施方案2中所述的化合物5、mtor抑制剂everolimus(依维莫司)以及imid免疫调节剂thalidomide(沙利度胺)/revlimid(来那度胺)/pomalidomide(泊马度胺)/cc-122/cc-220。
19.具体实施方案15所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、mtor抑制剂rapamycin(雷帕霉素)以及imid免疫调节剂thalidomide(沙利度胺)/revlimid(来那度胺)/pomalidomide(泊马度胺)/cc-122/cc-220。
20.具体实施方案15所述的三合一药物组合物为具体实施方案2中所述的化合物3、mtor抑制剂rapamycin(雷帕霉素)以及imid免疫调节剂thalidomide(沙利度胺)/revlimid(来那度胺)/pomalidomide(泊马度胺)/cc-122/cc-220。
21.具体实施方案15所述的三合一药物组合物为具体实施方案2中所述的化合物5、mtor抑制剂rapamycin(雷帕霉素)以及imid免疫调节剂thalidomide(沙利度胺)/revlimid(来那度胺)/pomalidomide(泊马度胺)/cc-122/cc-220。
22.一种三合一的药物组合物,其中含有btk抑制剂、mtor抑制剂以及bcl-2抑制剂。
23.具体实施方案22所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、mtor抑制剂everolimus(依维莫司)以及bcl-2抑制剂abt-199。
24.具体实施方案22所述的三合一药物组合物为具体实施方案2中所述的化合物3、mtor抑制剂everolimus(依维莫司)以及bcl-2抑制剂abt-199。
25.具体实施方案22所述的三合一药物组合物为具体实施方案2中所述的化合物5、mtor抑制剂everolimus(依维莫司)以及bcl-2抑制剂abt-199。
26.具体实施方案22所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、mtor抑制剂rapamycin(雷帕霉素)以及bcl-2抑制剂abt-199。
27.具体实施方案22所述的三合一药物组合物为具体实施方案2中所述的化合物3、mtor抑制剂rapamycin(雷帕霉素)以及bcl-2抑制剂abt-199。
28.具体实施方案22所述的三合一药物组合物为具体实施方案2中所述的化合物5、mtor抑制剂rapamycin(雷帕霉素)以及bcl-2抑制剂abt-199。
29.一种三合一的药物组合物,其中含有btk抑制剂、bcl-2抑制剂以及pi3k抑制剂。
30.具体实施方案29所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、bcl-2抑制剂abt-199以及pi3k抑制剂idelalisib。
31.具体实施方案29所述的三合一药物组合物为btk抑制剂具体实施方案2中所述的化合物3、bcl-2抑制剂abt-199以及pi3k抑制剂idelalisib。
32.具体实施方案29所述的三合一药物组合物为btk抑制剂具体实施方案2中所述的化合物5、bcl-2抑制剂abt-199以及pi3k抑制剂idelalisib。
33.具体实施方案29所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、bcl-2抑制剂abt-199以及pi3k抑制剂duvelisib。
34.具体实施方案29所述的三合一药物组合物为btk抑制剂具体实施方案2中所述的化合物3、bcl-2抑制剂abt-199以及pi3k抑制剂duvelisib。
35.具体实施方案29所述的三合一药物组合物为btk抑制剂具体实施方案2中所述的化合物5、bcl-2抑制剂abt-199以及pi3k抑制剂duvelisib。
36.一种三合一的药物组合物,其中含有btk抑制剂、mtor抑制剂以及methotrexate(甲氨蝶呤)。
37.具体实施方案36所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、mtor抑制剂everolimus(依维莫司)以及methotrexate(甲氨蝶呤)。
38.具体实施方案36所述的三合一药物组合物为具体实施方案2中所述的化合物3、mtor抑制剂everolimus(依维莫司)以及methotrexate(甲氨蝶呤)。
39.具体实施方案36所述的三合一药物组合物为具体实施方案2中所述的化合物5、mtor抑制剂everolimus(依维莫司)以及methotrexate(甲氨蝶呤)。
40.具体实施方案36所述的三合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)、mtor抑制剂rapamycin(雷帕霉素)以及methotrexate(甲氨蝶呤)。
41.具体实施方案36所述的三合一药物组合物为具体实施方案2中所述的化合物3、mtor抑制剂rapamycin(雷帕霉素)以及methotrexate(甲氨蝶呤)。
42.具体实施方案36所述的三合一药物组合物为具体实施方案2中所述的化合物5、mtor抑制剂rapamycin(雷帕霉素)以及methotrexate(甲氨蝶呤)。
43.一种二合一的药物组合物,其中含有btk抑制剂和mtor抑制剂。
44.具体实施方案43所述的二合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)和mtor抑制剂everolimus(依维莫司)。
45.具体实施方案43所述的二合一药物组合物为具体实施方案2中所述的化合物3和mtor抑制剂everolimus(依维莫司)。
46.具体实施方案43所述的二合一药物组合物为具体实施方案2中所述的化合物5和mtor抑制剂everolimus(依维莫司)。
47.具体实施方案43所述的二合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)和mtor抑制剂rapamycin(雷帕霉素)。
48.具体实施方案43所述的二合一药物组合物为具体实施方案2中所述的化合物3和mtor抑制剂rapamycin(雷帕霉素)。
49.具体实施方案43所述的二合一药物组合物为具体实施方案2中所述的化合物5和mtor抑制剂rapamycin(雷帕霉素)。
50.一种二合一的药物组合物,其中含有btk抑制剂和topk抑制剂。
51.具体实施方案50所述的二合一药物组合物为btk抑制剂ibrutinib(依鲁替尼)和topk抑制剂ots964。
52.具体实施方案50所述的二合一药物组合物为具体实施方案2中所述的化合物3和topk抑制剂ots964。
53.具体实施方案50所述的二合一药物组合物为具体实施方案2中所述的化合物5和topk抑制剂ots964。
54.一种二合一的药物组合物,其中含有pi3k抑制剂和topk抑制剂。
55.具体实施方案54所述的二合一药物组合物为pi3k抑制剂idelalisib和topk抑制剂ots964。
56.具体实施方案54所述的二合一药物组合物为pi3k抑制剂duvelisib和topk抑制剂ots964。
57.一种二合一的药物组合物,其中含有具体实施方案2中所述的化合物3和pi3k抑制剂idelalisib。
58.一种二合一的药物组合物,其中含有具体实施方案2中所述的化合物5和pi3k抑制剂idelalisib。
59.一种二合一的药物组合物,其中含有具体实施方案2中所述的化合物3和pi3k抑制剂duvelisib。
60.一种二合一的药物组合物,其中含有具体实施方案2中所述的化合物5和pi3k抑制剂duvelisib。
61.一种二合一的药物组合物,其中含有具体实施方案2中所述的化合物3和bcl-2抑制剂abt-199。
62.一种二合一的药物组合物,其中含有具体实施方案2中所述的化合物5和bcl-2抑制剂abt-199。
63.具体实施方案15-62的药物组合物在制备治疗或抑制癌症或其他疾病的药物中的用途,其中所述癌症或其他疾病包括,但不限于各种b细胞恶性肿瘤(包括小淋巴细胞淋巴瘤(sll)、慢性淋巴细胞白血病(cll)、弥漫性大b细胞淋巴瘤(dlbcl)、华氏巨球蛋白血症(waldenstrommacroglobulinemia)、滤泡性淋巴瘤(follicularlymphoma)、套细胞淋巴瘤(mcl)和多发性骨髓瘤(mm))以及其他抑制btk激酶活性对病人有益处的疾病。
64.具体实施方案15-62的药物组合物在制备治疗或抑制自身免疫性疾病(autoimmunediseases)的药物中的用途,所述自身免疫性疾病包括,但不限于器官特异性自身免疫疾病,如慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasturesyndrome)、寻常天皰疮、类天皰疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等,系统性自身免疫疾病,如系统性红斑狼疮、类风湿关节炎、系统性脉管炎、硬皮病、天疱疮、混合结缔组织病、自身免疫性溶血性贫血、甲状腺自身免疫疾病、溃疡性结肠炎等。
65.具体实施方案64的用途,其中所述自身免疫性疾病选自类风湿关节炎。
66.具体实施方案15-62的药物组合物在制备治疗或抑制异常免疫性疾病的药物中的用途,所述异常免疫性疾病包括,但不限于血清病、哮喘、过敏性鼻炎和药物过敏等。
67.具体实施方案15-62的药物组合物在制备治疗或抑制炎性疾病的药物中的用途,所述炎性疾病包括,但不限于角膜炎、鼻炎、口腔炎、腮腺炎、咽炎、扁桃体炎、气管炎、支气管炎、肺炎、心肌炎、胃炎、肠胃炎、胆囊炎、阑尾炎等。
68.具体实施方案15-62的药物组合物治疗或抑制癌症或其他疾病的方法,其中所述癌症或其他疾病包括,但不限于各种b细胞恶性肿瘤(包括小淋巴细胞淋巴瘤(sll)、慢性淋巴细胞白血病(cll)、弥漫性大b细胞淋巴瘤(dlbcl)、华氏巨球蛋白血症(waldenstrommacroglobulinemia)、滤泡性淋巴瘤(follicularlymphoma)、套细胞淋巴瘤(mcl)和多发性骨髓瘤(mm))以及其他抑制btk激酶活性对病人有益处的疾病。
69.具体实施方案15-62的药物组合物治疗或抑制自身免疫性疾病(autoimmunediseases)的方法,所述自身免疫性疾病包括,但不限于器官特异性自身免疫疾病,如慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasturesyndrome)、寻常天皰疮、类天皰疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等,系统性自身免疫疾病,如系统性红斑狼疮、类风湿关节炎、银屑病(psoriasis)、系统性脉管炎、硬皮病、天疱疮、混合结缔组织病、自身免疫性溶血性贫血、甲状腺自身免疫疾病、溃疡性结肠炎等。
70.具体实施方案68的方法,其中所述自身免疫性疾病选自类风湿关节炎。
71.具体实施方案15-62的药物组合物治疗或抑制异常免疫性疾病的方法,所述异常免疫性疾病包括,但不限于血清病、哮喘、过敏性鼻炎和药物过敏等。
72.具体实施方案15-62的药物组合物治疗或抑制炎性疾病的药物中的的方法,所述炎性疾病包括,但不限于角膜炎、鼻炎、口腔炎、腮腺炎、咽炎、扁桃体炎、气管炎、支气管炎、肺炎、心肌炎、胃炎、肠胃炎、胆囊炎、阑尾炎等。
- 还没有人留言评论。精彩留言会获得点赞!