一种目标区域捕获方法、试剂盒及测序方法与流程
1.本技术涉及目标区域捕获技术领域,特别是涉及一种目标区域捕获方法、试剂盒及测序方法。
背景技术:
2.目标区域测序(target region sequencing)是通过定制感兴趣的基因组区域的探针,与基因组dna进行杂交,将目标区域dna富集后进行高通量测序的研究策略。通过对大量样本的目标区域研究,有助于发现和验证疾病相关候选基因或相关位点,在临床诊断和药物开发方面有着巨大的应用潜力。杂交捕获的原理是人为设计dna或者rna探针,探针可以和目标区段部分或者全部互补;将样本和探针混合,探针会将目标区段捕获,未设计探针的区段会被洗脱丢弃;之后通过变性,一般是调节ph值到碱性,将探针和捕获区段分开;被捕获的片段即可进行二代测序文库构建。目标区域捕获适用于疾病的大样本量分析,包括帕金森、智力障碍、扩张型心肌病、乳腺癌等多种疾病的研究。
3.目标区域测序的目标区域捕获技术主要是,在溶液中,目标dna片段和带有生物素标记探针直接杂交,然后通过生物素亲和素的反应使目标dna片段锚定在带有亲和素的微珠上;洗去非目标dna,洗脱后,富集的dna用于测序。现有的目标区域捕获技术,都是针对短读长测序的二代测序平台开发和设计。包括pacbio官方和其他公司,在进行目标区域测序时,都是在二代测序的基础之上进行三代捕获测序技术的开发,由于方法的局限性,捕获效率极低,而且捕获到的基本都是短片段并没有充分利用第三代测序长读长的优点。
4.例如pacbio的目标区域测序,其目标区域捕获建库测序技术包括,起始样品为人血液中提取得到的总dna,g-tube打断后使用kapa hyper prep kits for illumina sequencing试剂盒加接头,pcr扩增和富集总基因组dna,使用idt生物素标记的rna探针捕获目的区域后,进行第二次pcr扩增富集目的区域,捕获pcr产物经损伤修复,末端修复,与已知接头smrt-bell连接,酶反应消化,bluepippin分选,最终得到哑铃形的文库,经过agilent2100和qubit hs检测合格后进行pacbio上机测序,数据下机后进行ccs数据校正。整个流程详细如下:
5.(1)设计探针,探针设计原则为:目的区域核酸序列,且与宿主基因组比对率低,长度为50nt的rna类型idt探针;
6.(2)将基因组dna打断至1-5kb;
7.(3)用kapa hyper prep kits for illumina sequencing试剂盒进行末端修复、加a、加y字形接头,即pacbio核酸序列;
8.(4)根据y字形接头上的primer,对基因组dna进行pcr扩增;
9.(5)样本混合pooling;
10.(6)使用步骤(1)设计好的特定探针捕获捕获目标区域核酸序列;
11.(7)m-270磁珠捕获目的区域;
12.(8)pcr扩增富集捕获到的目标区域核酸序列;
13.(9)损伤修复、末端修复、加smrt-bell接头、酶iii酶vii消化,均使用pacbio公布核酸序列;
14.(10)制备上机:加primer、加测序聚合酶;
15.(11)数据拆分:ccs校准。
16.以上在二代测序的基础之上开发的三代捕获测序技术,存在以下不足:
17.a、试验生产流程长,浪费人力物力成本较多;
18.b、建库过程中需要进行两次pcr扩增,然后上机测序,测序数据dup较高,造成较多的数据浪费;
19.c、由于建库过程中存在pcr过程,所以甲基化信息会在pcr过程中丢失,无法检测目标区域的甲基化修饰;
20.d、由于探针长度的限制,能够捕获的目的区域长度一般为1-5kb,无法发挥第三代测序长读长的优点。
技术实现要素:
21.本技术的目的是提供一种新的目标区域捕获方法、试剂盒及测序方法。
22.本技术具体采用了以下技术方案:
23.本技术的第一方面公开了一种目标区域捕获方法,包括根据目标区域核酸序列设计向导核酸序列,将向导核酸序列与cas9酶组合,采用向导核酸序列与cas9酶的复合物对待处理核酸样品进行酶切消化,回收酶切消化产物中符合设计的酶切消化的目标区域核酸序列长度的核酸片段,即捕获得到目标区域。
24.需要说明的是,本技术的目标区域捕获方法,利用crispr/cas9系统对目标区域进行酶切,获得预期大小的目标区域核酸片段,然后采用核酸片段分离的方法,获取目标区域。本技术的目标区域捕获方法,流程短,不仅节省了交付周期和人力成本;而且,提升了捕获效率,增加了有效数据的利用率,节约了测序成本;并且,本技术的捕获方法整个捕获过程中无需采用pcr扩增,可以保留基因组dna原始修饰信息,直接检测目标区域甲基化等信息。
25.还需要说明的是,本技术的目标区域捕获方法,不仅适用于二代测序,更适用于第三代测序,打破了现有技术普遍在第二代测序的基础之上进行第三代测序目标区域捕获技术开发的技术限制,解决了针对目标区域的第三代测序受第二代测序捕获技术限制的技术问题。本技术的目标区域捕获方法,其捕获的目标区域产物可以用于任何测序或检测技术;例如,加入不同的测序接头就可以适用于不同的测序平台。
26.优选的,本技术的目标区域捕获方法中,向导核酸序列由crrna引物与tracrrna引物退火组装形成。
27.可以理解,在crispr/cas9系统中,向导核酸序列是由设计的crrna引物与通用的tracrrna引物退火组装形成;其中,设计的crrna引物即针对目标区域设计的精确靶向目标区域的寡核苷酸;而tracrrna引物则是一段与设计的crrna引物退火形成发夹结构的寡核苷酸核酸序列,crrna引物与tracrrna引物退火组装成向导核酸序列。
28.优选的,本技术的目标区域捕获方法,还包括对酶切消化产物进行磁珠纯化,然后再进行核酸片段筛选,捕获目标区域。
29.需要说明的是,磁珠纯化的目的是为了方便后续核酸片段筛选,可以理解,除了磁珠纯化以外,不排除还可以采用其它的核酸纯化方法。
30.优选的,本技术的目标区域捕获方法中,回收酶切消化产物中的核酸片段,具体采用blue pippin自动化核酸回收系统进行目标区域的核酸片段分选。
31.需要说明的是,blue pippin只是本技术的一种实现方式中具体采用的核酸回收系统,不排除还可以采用其它分离不同大小核酸片段的方法或系统。
32.本技术的第二方面公开了一种目标区域捕获试剂盒,包括针对目标区域核酸序列设计的crrna引物和通用的tracrrna引物。
33.需要说明的是,本技术的目标区域捕获方法,其关键在于针对目标区域设计crrna引物和通用的tracrrna引物;因此,本技术的一种实现方式中,将设计的crrna引物和通用的tracrrna引物作为一个试剂盒,以方便使用。可以理解,本技术目标区域捕获方法中使用的其它试剂,可以自行购买获得,或者将其全部或部分组合到本技术的试剂盒中,以方便使用。
34.因此,优选的,本技术的试剂盒中,还包括cas9酶、crrna引物与tracrrna引物退火组装试剂、向导核酸序列与cas9酶组合试剂、cas9酶的酶切消化试剂和磁珠纯化试剂中的至少一种。
35.本技术的第三方面公开了一种测序方法,包括采用本技术的目标区域捕获方法或者本技术的目标区域捕获试剂盒进行目标区域捕获,捕获的目标区域核酸片段直接用于测序文库构建和测序。
36.需要说明的是,本技术的测序方法,不是在二代测序的基础之上开发捕获目标区域,因此,没有二代测序方法的局限性。本技术的测序方法,由于采用本技术的目标区域捕获方法或试剂盒,原则上可以获得很长的目标区域核酸片段,这尤其适用于长读长的测试方法,例如第三代测序,能充分发挥第三代测序长读长的优点。本技术测序方法不仅能提升捕获效率,增加有效数据利用率,节约测序成本;而且,可以保留基因组dna原始修饰信息,实现目标区域甲基化等信息检测和分析。
37.优选的,本技术的测序方法中,测序文库构建采用pacbio建库。
38.需要说明的是,采用pacbio建库是因为本技术的一种实现方式中具体采用的是第三代测序平台中的pacbio平台。可以理解,如果采用不同的测序平台,测序文库构建方案也可以进行相应的调整和改变,具体参考各测序平台的使用说明。例如,本技术的目标区域捕获方法和试剂盒,也可以在ont纳米孔测序平台使用;因此,测序文库构建也可以参考ont纳米孔测序平台进行。
39.本技术的第四方面公开了一种目标区域甲基化检测方法,包括采用本技术的测序方法对目标区域进行测序,根据测序结果分析目标区域的甲基化情况。
40.需要说明的是,本技术的测序方法,其最大的特点即,在进行目标区域捕获时无需采用pcr,可以保留目标区域的原始修饰信息,包括甲基化等信息。因此,本技术的测序方法也可以用于目标区域的甲基化检测。
41.本技术的第五方面公开了一种人atxn10基因捕获试剂盒,包括atxn10基因特异性crrna引物和通用的tracrrna引物;atxn10基因特异性crrna引物包括seq id no.1所示核酸序列的寡核苷酸片段和seq id no.2所示核酸序列的寡核苷酸片段;
42.seq id no.1:5
’-
tgttccaccagcctttgcca-3’43.seq id no.2:5
’-
taaatttcacctgatcaagg-3’。
44.需要说明的是,人atxn10基因捕获只是本技术的一种实现方式中为了验证本技术的目标区域捕获方法,特别设计的实验方案。可以理解,基于本技术的发明思路,不仅可以用于人atxn10基因捕获,也可以用于其它目标区域的捕获。并且,seq id no.1和seq id no.2所示核酸序列的atxn10基因特异性crrna引物,也只是本技术的一种实现方式中具体采用的引物;在本技术发明构思基础上,还可以针对atxn10基因设计不同的引物,在此不作具体限定。
45.还需要说明的是,在设计获得特异性crrna引物的基础上,tracrrna引物可以根据crispr/cas9系统的常规设计方案获得;例如本技术的一种实现方式中,在设计了atxn10基因特异性crrna引物的基础上,tracrrna引物的功能核酸序列为idt公司提供的通用核酸序列,该通用核酸序列参考试剂盒universal sequence providedby idt:1072532。
46.优选的,本技术的人atxn10基因捕获试剂盒中,还包括cas9酶、atxn10基因特异性crrna引物与tracrrna引物退火组装试剂、向导核酸序列与cas9酶组合试剂、cas9酶的酶切消化试剂和磁珠纯化试剂中的至少一种。
47.本技术的有益效果在于:
48.本技术的目标区域捕获方法,流程短、操作简单,节省了交付周期和人力成本;并且,捕获效率高,增加了有效数据的利用率,节约了测序成本。本技术打破了现有的目标区域捕获普遍基于第二代测序技术基础开发的技术限制,创造性的利用crispr/cas9系统进行目标区域捕获,整个捕获过程无需采用pcr扩增,能够保留目标区域基因的甲基化等原始修饰信息。
附图说明
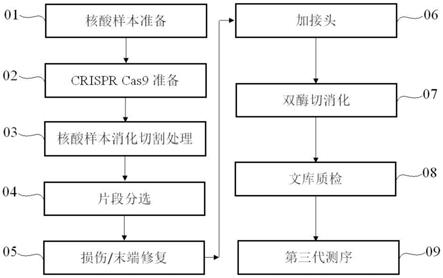
49.图1是本技术实施例中目标区域捕获方法及建库测序的流程框图。
具体实施方式
50.目前的实践中,在将目标区域捕获分离时,都是针对短读长测序的二代测序平台开发和设计的目标区域捕方案。这些方案不仅捕获效率低、流程长、成本高、测序数据利用率低;而且捕获到的基本都是短片段,不能发挥长读长测序方法(例如第三代测序)的优点;更重要的是,整个捕获过程需要进行pcr扩增,这会导致目标区域的甲基化等原始修饰信息丢失。
51.crispr/cas系统是一种原核生物的免疫系统,用来抵抗外源遗传物质的入侵,比如噬菌体病毒和外源质粒。同时,它为细菌提供了获得性免疫:这与哺乳动物的二次免疫类似,当细菌遭受病毒或者外源质粒入侵时,会产生相应的“记忆”,从而可以抵抗它们的再次入侵。crispr/cas系统可以识别出外源dna,并将它们切断,沉默外源基因的表达。正是由于这种精确的靶向功能,crispr/cas系统被开发成一种高效的基因编辑工具。在自然界中,crispr/cas系统拥有多种类别,其中crispr/cas9系统是研究最深入,应用最成熟的一种类别。凭借着成本低廉,操作方便,效率高等优点,crispr/cas9迅速风靡全球的实验室,成为生物科研的有力帮手。
52.基于以上研究和认识,本技术创造性的将crispr/cas9系统引入目标区域捕获,利用crispr/cas9系统精确靶向切割功能,针对目标区域核酸序列设计crrna引物和通用的tracrrna引物,对目标区域进行精确切割,然后根据切割后的片段大小,回收目标区域,实现目标区域捕获。
53.本技术的一种实现方式中,整个目标区域捕获和测序,如图1所示,包括以下步骤:
54.(1)核酸样本准备01步骤,该步骤主要是对提取的核酸样本进行质检,要求其总量大于5μg,片段长度需要大于40kb,且没有拖尾现象发生。
55.(2)crispr cas9准备02步骤,即将设计的crrna引物和通用的tracrrna引物组装形成guide dnaoligo,即向导核酸序列,再与cas9酶组合形成酶复合物;其中,crrna引物是每隔一定距离设计一个切割位点,使需要捕获的目标区域长度在1-35kb之间;这个长度可以很好的发挥第三代测序的长读长优点。
56.(3)核酸样本消化切割处理03步骤,即将guide dna oligo与cas9酶的复合物加入到核酸样本中进行消化处理;在本技术的一种实现方式中,进一步的还包括,对消化处理的产物进行磁珠纯化。
57.(4)片段分选04步骤,本技术的一种实现方式中,具体采用blue pippin片段分选目标区域,按照目标区域的长度,选择合适的切割范围;比如,本实施例捕获基因长度为5kb,可以在上下游1kb进行切割,即切割范围为4-6kb,据此设计crrna引物,然后按照该范围分选核酸片段即可捕获得到目标区域;
58.(5)常规pacbio建库,主要包括损伤/末端修复05步骤、加接头06步骤、双酶切消化07步骤,可以参考现有的常规pacbio建库。
59.(6)文库质检08步骤,主要是对构建的测序文库进行质检,判断其是否符合测序要求,按照常规的测序文库质检方法即可。
60.(7)第三代测序09步骤,即采用第三代测序平台进行测序,本技术的一种实现方式中,具体采用的是pacbio平台进行测序,原则上,本技术的目标区域捕获方法也可以适用于其它的测序平台,例如ont纳米孔测序平台,相应的建库步骤也需要按照测序平台要求进行调整,在此不作具体限定。
61.本技术的捕获方法捕获效率高、流程短、成本低、测序数据利用率高;而且目标区域片段大小可以根据要求进行设计,整个捕获过程无需进行pcr扩增,能够很好的保留目标区域的甲基化等原始修饰信息。将本技术的目标区域捕获方法用于第三代测序,能够很好的发挥第三代测序长读长的优点。当然,本技术的目标区域捕获方法不仅限用于第三代测序,也能够用于其它测序或检测。
62.下面通过具体实施例对本技术作进一步详细说明。以下实施例仅对本技术进行进一步说明,不应理解为对本技术的限制。
63.实施例
64.本例使用人标准品na12878,使用crispr/cas9系统捕获22号染色体上面一段attct串联重复核酸序列,即atxn10基因。首先,设计atxn10捕获需要的crrna引物和tracrrna引物核酸序列,将crrna引物和tracrrna引物退火后与cas9酶进行组装,然后用于酶切消化核酸分子上面的目的区域。最后,使用blue pippin进行片段分选,筛选出目的长度的片段即为捕获产物。本例进一步的,对捕获产物进行了pacbio建库测序和测序结果分
析。详细如下:
65.1.样本准备的准备
66.本例采用人标准品na12878基因组dna进行试验。如果从全血样品中提取核酸,则需要对提取的核酸进行质检,片段长度需要在40kb以上,且没有拖尾现象发生,总量大于5μg,且样本无色、澄清、透明,才可用于后续的crispr处理和建库测序。
67.2.crispr-cas9引物准备
68.(1)引物设计
69.本例针对atxn10基因设计了两条atxn10基因特异性crrna引物,分别命名为atxn10.dc.1和atxn10.dc.2,atxn10.dc.1为seq id no.1所示核酸序列,atxn10.dc.2为seq id no.2所示核酸序列。两条crrna引物均由idt公司合成,并采用idt公司提供的通用核酸序列合成本例通用的tracrrna引物,即试剂盒universal sequence provided by idt:1072532。两条atxn10基因特异性crrna引物如表1所示。
70.表1atxn10基因特异性crrna引物
[0071][0072]
本例在以下网站设计atxn10的crrna引物核酸序列:
[0073]
https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design。
[0074]
(2)引物稀释
[0075]
使用纯水稀释引物至50μm储存备用,稀释后-20℃储存。
[0076]
(3)引物退火
[0077]
首先将引物按照表2的反应体积配制,然后95℃反应5min,然后将反应体系放置在室温自然冷却,即完成crrna引物和tracrrna引物退火。退火后的引物命名为guide dna oligo,即向导核酸序列。
[0078]
表2引物退火体系
[0079]
试剂名称储存浓度使用体积atxn10.dc.1或atxn10.dc.250μm1μltracrrna50μm1μlnuclease-free duplex buffer-8μl
[0080]
3.cas9 digestion酶消化
[0081]
本例采用vazyme cas9 nuclease酶(lot:en301-01/02)试剂盒进行酶切消化,详细如下:
[0082]
(1)cas9酶和guide dna oligo的组合:按照表3配制反应体系,混匀离心后在37℃孵育10min后,置于冰上备用。
[0083]
表3 guide dna oligo与cas9酶组合体系
[0084]
试剂名称储存浓度使用体积
nebuffer3.110
×
2.0μlguide dna oligo5μm8.0μlcas9 nuclease20μm2.0μlwater(nuclease-free)-8.0μl
[0085]
(2)目标区域消化:将cas9酶和guide dnaoligo的复合物,加入到80μl的核酸中,混匀离心后在37℃孵育60min后,置于冰上备用。
[0086]
(3)cas9失活处理:加入5μl浓度为0.5m的edta,混匀离心后放置在冰上备用。
[0087]
(4)消化产物的磁珠纯化:本例采用诺唯赞vahtstm dna clean beads(lot:n411)试剂盒进行磁珠纯化。具体的,加入100μl的ampure pb beads磁珠到样本中,结合10min后,用75%的无水乙醇清洗两次,最后用30μl的水将核酸从磁珠上回溶下来。
[0088]
4.片段筛选
[0089]
使用blue pippin筛选4-6kb之间的目的长度核酸分子;本例的捕获基因长度为5kb,根据设计,在目标区域上下游1kb进行切割,即切割范围为4-6kb,因此本例筛选4-6kb之间的目的长度核酸分子,即可捕获得到目标区域。
[0090]
5.pacbio建库
[0091]
(1)损伤修复反应
[0092]
本例采用pacbio建库试剂盒smrtbell template prep kit 1.0(lot:100-259-100)进行测序文库构建。其中,损伤修复采用m6630l nebnext ffpe dna repair mix 96 reaction neb(neb,1000002572)试剂盒。
[0093]
本例的损伤修复按照表4配制反应体系,然后,在37℃反应60分钟,进行损伤修复。
[0094]
表4损伤修复反应体系
[0095]
试剂用量(μl)dna damage repair buffer5nad+0.5atp high5dntp0.5dna damage repair mix1dna(片段筛选产物)38
[0096]
(2)末端修复反应
[0097]
将损伤修复反应产物50μl与2μl的dna damage repair mix混合,然后,在25℃反应10分钟,进行末端修复。
[0098]
(3)加接头
[0099]
按照表5配制加接头反应体系,然后,25℃反应12~16小时,本例具体反应12小时,进行接头连接。
[0100]
表5加接头反应体系
[0101]
试剂用量(μl)末端修复的dna20annealed blunt adapter(20um)10template prep buffer4
atp low2ligase1h2o3
[0102]
(4)双酶消化
[0103]
利用exoiii和exovii对加接头产物进行酶消化处理,酶消化处理的反应体系如表6所示。配制好反应体系后,在37℃酶消化60分钟。
[0104]
表6酶消化处理反应体系
[0105]
试剂用量(μl)加接头的dna40exoiii1exovii1
[0106]
6.上机测序
[0107]
本例采用三代测序平台中的pacbio平台进行测序。
[0108]
数据下机后进行ccs数据校正,进行下机数据量、下机reads数、有效reads数、捕获效率、duplicate rates和m6a甲基化修饰程度统计和分析。
[0109]
对比试验
[0110]
本例采用现有的pacbio目标区域捕获建库测序技术对相同的人标准品na12878,进行相同的atxn10基因捕获、建库和测序。pacbio目标区域捕获建库测序技术,对人标准品na12878基因组dna进行g-tube打断后使用kapa hyperprep kits for illumina sequencing试剂盒加接头,pcr扩增和富集总基因组dna,使用idt生物素标记的rna探针捕获目的区域后,进行第二次pcr扩增富集目的区域,第二次pcr扩增产物经损伤修复、末端修复,与已知接头smrt-bell连接,酶反应消化,bluepippin分选,最终得到哑铃形的文库,经过agilent2100和qubit hs检测合格后进行pacbio上机测序,数据下机后进行ccs数据校正。详细如下:
[0111]
1.探针设计
[0112]
针对atxn10基因设计目的区域带生物素标记的rna探针,探针设计原则为:长度为50nt的rna类型idt探针。探针设计和合成由idt公司完成。
[0113]
2.基因组dna打断
[0114]
本例采用g-tube,13000rpm/min的打断参数将基因组dna打断至1-5kb。
[0115]
3.末端修复、加a和加y字形接头
[0116]
本例采用kapa hyper prep kits for illumina sequencing试剂盒进行末端修复和加a,y字形接头为pacbio核酸序列。
[0117]
(1)末端修复和加a
[0118]
按照表7的反应体系配制末端修复和加a的反应体系,然后在pcr反应仪中进行20℃保温30min、65℃保温30min,4℃待机。
[0119]
表7端修复和加a反应体系
[0120]
成分用量(μl)sheared dna50end repair&a-tailing buffer7
end repair&a-tailing enzyme mix3总体积60
[0121]
(2)y字形接头连接
[0122]
按照表8的反应体系配制y字形接头连接反应体系,然后,20℃,15min,进行加接头反应。
[0123]
表8加接头反应体系
[0124]
成分体积(μl)end repair&a-tailing reaction product60pcr-grade water5ligation buffer30dna ligase10annealed barcoded adapter5
[0125]
4.第一次pcr扩增
[0126]
根据y字形接头上的primer对基因组dna进行pcr扩增,pcr扩增反应体系参考y字形接头为pacbio核酸序列的基因组扩增,pcr扩增反应条件为:95℃变性2min,然后进入6个循环:95℃20seconds、62℃20seconds、68℃10min,循环结束后68℃5min,4℃hold。
[0127]
第一次pcr扩增完成后,采用idt公司提供的atxn10基因的带生物素标记的探针,对atxn10基因进行目标区域核酸序列捕获,详细如下:
[0128]
a、加5μl的cot human dna(1mg/ml)到一个1.5ml离心管中;
[0129]
b、加2.0μg前面加上接头的文库,即第一次pcr扩增产物,到上一步骤的离心管中;
[0130]
c、加10μl引物到上一步骤离心管中并混匀离心,该引物即pacbio y字型接头上面的引物核酸序列,目的是在杂交前让该引物退火到y字形接头上面去,在探针捕获方法中用来减少非特异性捕获;
[0131]
d、使用浓缩仪将上述样本浓缩为干粉;
[0132]
e、准备杂交反应试剂:xgen 2x hyb buffer 8.5μl、xgen hyb buffer enhancer2.7μl、水1.8μl;
[0133]
f、将杂交反应试剂加入到浓缩干粉中,混匀离心后,95℃孵育10分钟后;
[0134]
g、加入4μl的探针,混匀、离心后,放在65℃孵育4个小时。
[0135]
5.链酶亲和素磁珠的洗脱准备
[0136]
磁珠室温孵育30分钟以上,使用200μl的wash buffer i清洗两次磁珠。然后使用100μl的wash buffer i回融磁珠。
[0137]
6.目标区域捕获
[0138]
采用等体积的磁珠,对探针与加接头文库的孵育产物进行结合,结合条件为室温45min;然后,使用1
×
stringent wash buffer清洗一次,放在65℃孵育10分钟;使用1
×
wash buffer i清洗一次,放在65℃孵育10分钟;使用1
×
wash bufferii清洗一次,放在65℃孵育10分钟;使用1
×
wash buffer iii清洗一次,放在65℃孵育10分钟。
[0139]
第二次pcr扩增:经过1
×
wash buffer iii清洗后,直接对产物进行pcr扩增,即获得捕获富集的目标区域。
[0140]
第二次pcr扩增的反应体系如表9所示,pcr扩增反应条件为:95℃变性2min,然后
进入15个循环:95℃20seconds、62℃20seconds、68℃10min,循环结束后68℃5min,4℃hold。
[0141]
表9加接头反应体系
[0142]
成分体积(μl)captured library5010
×
la pcr buffer202.5mm each dntps16100μm pacbio universal primer2takara la taq dna polymerase1.2water110.8
[0143]
7.pacbio建库
[0144]
(1)损伤修复反应
[0145]
本例采用pacbio建库试剂盒smrtbell template prep kit 1.0(lot:100-259-100)进行测序文库构建。其中,损伤修复采用m6630l nebnext ffpe dna repair mix 96 reaction neb(neb,1000002572)试剂盒。
[0146]
本例的损伤修复按照表4配制反应体系,然后,在37℃反应60分钟,进行损伤修复。
[0147]
(2)末端修复反应
[0148]
将损伤修复反应产物50μl与2μl的dna damage repairmix混合,然后,在25℃反应10分钟,进行末端修复。
[0149]
(3)加接头
[0150]
按照表5配制加接头反应体系,然后,25℃反应12~16小时,本例具体反应12小时,进行接头连接。
[0151]
(4)双酶消化
[0152]
利用exoiii和exovii对加接头产物进行酶消化处理,酶消化处理的反应体系如表6所示。配制好反应体系后,在37℃酶消化60分钟。
[0153]
8.上机测序
[0154]
本例采用三代测序平台中的pacbio平台进行测序。
[0155]
数据下机后进行ccs数据校正,进行下机数据量、下机reads数、有效reads数、捕获效率、duplicate rates和m6a甲基化修饰程度统计和分析。
[0156]
本例对比分析了本例目标区域捕获方法的测序结果,和对比试验采用现有pacbio目标区域捕获建库测序的测序结果,如表10所示。
[0157]
表10测序结果对比
[0158]
[0159]
从表10的结果可以看出,无论是本例的目标区域捕获方法(以下简称“实验组”),还是对比试验的现有方法(以下简称“对照组”),数据产量均大于5gb,就数据产量来说,两种捕获方法,均未对测序结果造成不利的影响,说明本例的目标区域捕获方法未对测序结果造成任何不利的影响。
[0160]
关于重复序列比对到基因组上的比例duplicate rate on genome(%),相较于对照组的8.01%,实验组优化后降低至0.42%,可利用数据提高。说明采用实验组的方法可以明显的降低重复序列比,从而提高数据的利用率。
[0161]
关于甲基化修饰,对照组由于捕获过程中有pcr过程,丢失了修饰信息,所以未能检测出基因上面的m6a甲基化修饰信息。而本例采用crispr方法,是一种不需要pcr的捕获,所以保留了基因上面的甲基化修饰信息。
[0162]
从以上试验和对比结果可以看出,采用本例的目标区域捕获方法,减少了pcr的过程,减少重复序列(duplicate)产生的同时,还可以保留目标区域的甲基化等修饰信息,可以有效提高数据利用率,极大的节约测序成本。
[0163]
以上内容是结合具体的实施方式对本技术所作的进一步详细说明,不能认定本技术的具体实施只局限于这些说明。对于本技术所属技术领域的普通技术人员来说,在不脱离本技术构思的前提下,还可以做出若干简单推演或替换。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1