用于深海微生物原位细胞裂解的组合物、试剂、试剂盒及应用的制作方法
本发明涉及深海微生物领域,特别涉及深海原位细胞裂解组合物、裂解液、试剂盒及其应用。
背景技术:
深海生物在整个海洋甚至全球的生态系统中扮演着重要角色。采集不同深度海水中的生物样品,是现代海洋学研究的重要内容。随着各种海洋科学研究与海洋资源开发利用的不断深入,如何快速、方便和有效地对海洋生物进行调查采样,以获得第一手的海洋生物科学研究样品,全面了解特定海域的生物资源情况,成为了当务之急。和传统的采样分析相比,近年发展起来的深海原位细胞富集采样是最有效的深海生物核酸获取手段,是对传统海洋生物学研究方法的一次重大突破。
深海高压低温,如何在原位环境中使深海生物细胞裂解并提取核酸是深海微生物研究的难题。尤其是原位获得的高质量rna是研究深海生物基因功能的前提。深海核酸提取仪采用泵连接过滤腔(内含生物滤膜)的方式对海水大通量过滤。当过滤足量的海水体积后,触发转换阀切换管路,使泵的入口由海水切换为细胞裂解液,通过置换管路内原有的海水实现过滤腔内微生物和幼虫的细胞支撑蛋白裂解。收集的核酸可以用于高通量测序来研究深海生物的宏基因组和转录组。因此,深海原位富集细胞裂解液对于原位获得深海生物核酸具有重要的现实意义。
技术实现要素:
有鉴于此,本发明提供了用于深海微生物原位细胞裂解的组合物、试剂、试剂盒及应用。该方法可以在深海低温高压条件下直接获取海水中微生物的核酸。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了用于深海微生物原位细胞裂解的组合物,包括:tris-hcl25~50mm、nacl100~200mm、edta1~10mm、tritonx-1001~2%(v/v)、溶菌酶3~5mg/ml、sds0.05~0.1%(v/v)、蛋白酶k10~20μl/ml。
在本发明的一些具体实施方案中,组合物包括:tris-hcl50mm、nacl150mm、edta1mm、tritonx-1001%(v/v)、溶菌酶3mg/ml、sds0.05%、蛋白酶k20μl/ml。
在上述研究的基础上,本发明还提供了所述的组合物在制备深海微生物原位细胞裂解的试剂和/或试剂盒中的应用。
本发明还提供了所述的组合物在制备深海微生物原位核酸提取的试剂和/或试剂盒中的应用。
本发明还提供了所述的组合物在制备深海微生物核酸检测的试剂和/或试剂盒中的应用。
在上述研究的基础上,本发明还提供了深海微生物原位细胞裂解的试剂或试剂盒,包括所述的组合物以及可接受的助剂。
在上述研究的基础上,本发明还提供了深海微生物原位核酸提取的试剂或试剂盒,包括所述的组合物以及可接受的助剂。
在上述研究的基础上,本发明还提供了深海微生物核酸检测的试剂或试剂盒,包括所述的组合物以及可接受的助剂。
在上述研究的基础上,本发明还提供了深海微生物原位细胞裂解的方法,采用所述的组合物或所述的深海微生物原位细胞裂解的试剂或试剂盒。
在上述研究的基础上,本发明还提供了深海微生物原位核酸提取和/或检测的方法,采用所述的组合物或所述的深海微生物原位核酸提取的试剂或试剂盒或所述的深海微生物核酸检测的试剂或试剂盒。
本发明提供的一套可以适用于全海深的原位核酸提取及回收的方法,该方法有效的简化了实验室深海微生物样品的提取方式,使其可以置于深海中进行原位的提取;本发明中的配置的裂解液可以有效地裂解包括厚壁菌在内的深海细菌和微生物,获得与常规实验室使用试剂盒等量的核酸;与现有的深海采样技术相比较,该系统的采样流程可以有效的避免由于温度、压力等条件改变而影响原位核酸,也避免了实验室提取过程可能存在的污染;此外,本发明配合全海深微生物核酸原位提取装置可以完成在一次下潜采样中获得多达9个海水微生物的核酸样品。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
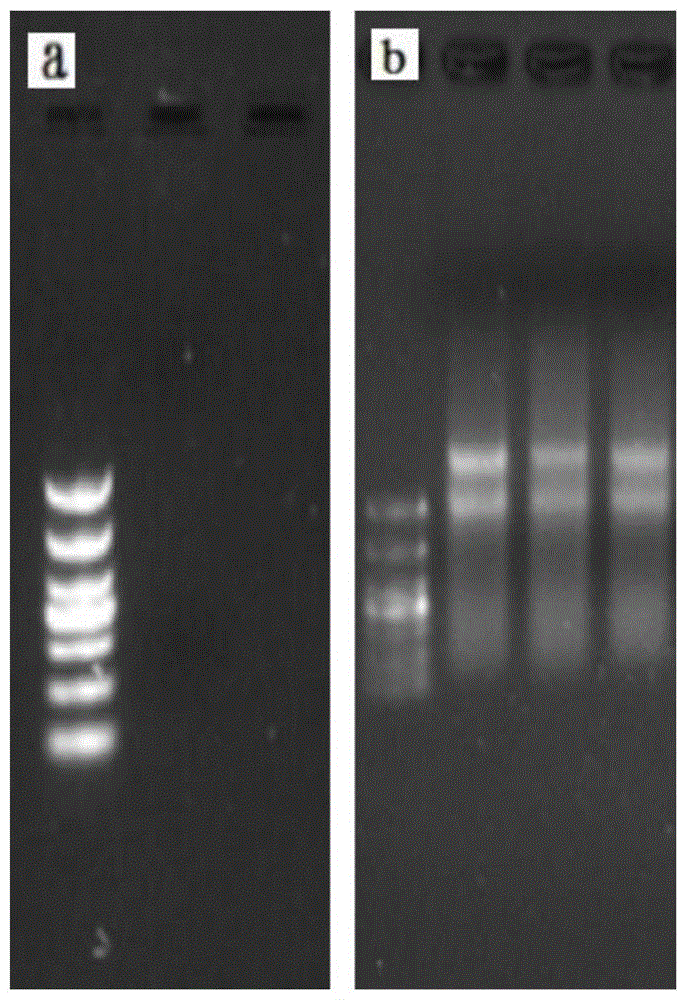
图1示rna电泳图;其中图1(a)为南海北部1000m水样rna电泳图,图1(b)为新鲜海水样品提取rna图;
图2示吸附条件对比实验流程图;
图3示第一次均匀液体替换实验;
图4示第二次均匀液体替换实验;
图5示第三次均匀液体替换实验;
图6示第一次盐溶液替换实验(单位:ppt);
图7示第二次盐溶液替换实验(单位:ppt);
图8示第一次原位富集装置-管路替换实验(单位:ppt);
图9示第二次原位富集装置-管路替换实验(单位:ppt);
图10示第三次原位富集装置-管路替换实验(单位:ppt);
图11示核酸低温保存实验。
具体实施方式
本发明公开了用于深海微生物原位细胞裂解的组合物、试剂、试剂盒及应用,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
为了成功在深海原位状态下获得海水中微生物的核酸,将提取过程分为三个步骤:微生物富集、细胞裂解和核酸回收。
1、微生物富集:使用海水泵将海水注入已经安装有0.22um孔径的滤膜,富集微生物于滤膜上。使用阀门切换的方式可以改变管路通向,配合全海深微生物核酸原位提取装置上的多个滤膜腔,可以一次完成9组微生物样品的富集工作。优选9个滤膜腔。
2、细胞裂解:针对深海高压低温的条件,本发明中配置了一种是可以适用于深海条件的细胞裂解液,有效地裂解深海生物细胞,获得高质量核酸。裂解的配方如下:tris-hcl(50mm):缓冲体系,提供合适的裂解环境;
nacl(150mm):等渗体系,维持核酸结构的稳定;
edta(1mm):变性剂和稳定剂,抑制样品中的核酸酶在裂解工程中对核酸的破坏;
tritonx-100(1%):非离子型表面活性剂,破坏细胞膜(裂解细胞)以释放细胞内的可溶性物质;
溶菌酶(3mg/ml):催化水解糖苷键,破坏细胞壁中的β-1,4糖苷键,使细胞壁不溶性黏多糖分解成可溶性糖肽,导致细胞壁破裂;
sds(0.05%):阴离子表面活性剂,溶解细胞膜上的脂质和蛋白从而破坏细胞;
蛋白酶k(20μl/ml):降解蛋白质,使蛋白质变小,促进蛋白质的游离,防止蛋白质和dna结合。
微生物富集完成后,通过阀门的切换,将海水泵管路切换为裂解液注入蠕动泵。向滤膜腔中注入裂解液置换滤膜腔内海水,并静置30min裂解细胞。裂解完成后,同时打开裂解液注入蠕动泵和装置后端的乙醇注入蠕动泵,混合后的液体用于后续核酸回收。
3、核酸回收:核酸回收方式选择使用吸附柱进行回收。将上述混合后的液体在蠕动泵的作用下,缓慢注入吸附柱中,混合液中的核酸会吸附于吸附柱上。配合全海深微生物核酸原位提取装置上的多个吸附柱,通过切换吸附柱阀门,即可一次完成多组微生物样品的核酸收集工作。优选9个吸附柱。
在深海原位状态下,配合全海深微生物核酸原位提取装置上的9个滤膜腔和对应的9个吸附柱,通过切换不同管路,打开不同泵和阀门,可以在一次下潜采样完成9组采样过程,包括微生物富集、裂解和核酸回收。
该方法可以在深海低温高压条件下直接获取海水中微生物的核酸。首先通过装置的海水泵将大量海水过滤到滤膜腔中,完成海水中微生物的富集;再通过蠕动泵将裂解液注入到滤膜腔,通过化学裂解方式破坏蛋白从而释放核酸;最后使用核酸吸附柱对核酸进行回收和保存;同时依靠深海核酸提取装置,可以一次获得多达9组的深海海水中微生物的核酸样品。本发明的技术方案中使用的裂解液与商业常温裂解液具有等同效用,且可以有效的避免传统采样方式中由于温度、压力等变化对核酸的影响。目前该技术方法已经成功用于全海深微生物核酸原位提取装置,获得微克级的核酸量。
本发明采用上述技术方案的优点是:
本发明提供的一套可以适用于全海深的原位核酸提取及回收的方法,该方法有效的简化了实验室深海微生物样品的提取方式,使其可以置于深海中进行原位的提取;本发明中的配置的裂解液可以有效地裂解包括厚壁菌在内的深海细菌和微生物,获得与常规实验室使用试剂盒等量的核酸;与现有的深海采样技术相比较,该系统的采样流程可以有效的避免由于温度、压力等条件改变而影响原位核酸,也避免了实验室提取过程可能存在的污染;此外,本发明配合全海深微生物核酸原位提取装置可以完成在一次下潜采样中获得多达9个海水微生物的核酸样品。
本发明提供的用于深海微生物原位细胞裂解的组合物、试剂、试剂盒及应用中所用原料及试剂均可由市场购得。
本发明提供的方法是属于深海原位状态下的实验,其技术效果的预期目标是可以获得的足够的用于测序的核酸量,现有的实验室操作结果可作为是作为参考值,不作为统计学比较的数值。本发明的最主要的技术效果是可以在深海原位状态下直接获取海水中微生物的核酸。
本发明是以深海原位宏基因组和宏转录组为主要研究内容,但是能够提供关于功能基因的位置和预测作用机理信息的信使rna极不稳定,死亡细胞的rna几分钟内即可解。如图1,深海微生物经过富集后实验室提取rna,发生了明显降解。但新鲜海水样品提取rna,仍然保持完整性,可以获得更多的原位信息。此外,由于深海生物量比较低,需要过滤大量的海水以便获得足够的生物量。当过滤足量海水后,通过使用细胞裂解液置换管路原有海水实现过滤腔内微生物的细胞壁或细胞膜裂解。
针对以上目的设计实验室验证实验如下:实验样品、裂解实验、吸附对比实验、吸附条件对比实验、替代效果检测、核酸保存实验。
实验样品:
实验样品选择深海研究所附近海域海水,每次实验开始前,使用在距离岸边15m的区域采集的新鲜水样,过滤0.2um滤膜(milliporetm,直径47mm),过滤体积是2l,过滤后将滤膜保存在无菌平板中。
使用天根dna/rna共提试剂盒(dp422),在实验室条件下提取,裂解时间为30min。得到dna总量最大值是560ng,rna总量最大值是620ng。
裂解实验:
有深海中温度低、压力大等条件会影响裂解液对微生物或幼虫的裂解效率,因此需要评估各个环境因子对裂解效率的影响,寻找最佳的处理流程,验证裂解液在低温条件下裂解效率。使用常用破坏细胞壁蛋白的解耦剂,包括sds、triton-x100、trizol等,分别作用到等量的纯培养环境细菌液、纯培养深海细菌、海水富集细菌。在实验室环境下,其他条件相同时比较提取核酸的效果。
下面结合实施例,进一步阐述本发明:
实施例1裂解液的筛选
a、使用实验室常用核酸提取裂解液:含1%sds或的碱性裂解液、0.1%sds或的碱性裂解液、1%triton碱性裂解液,以天根试剂盒(tiangendp302)提取作为标准。作用于过滤了等体积海水的滤膜上,裂解过程中将离线管至于4℃,裂解相同时间后,使用试剂盒回收dna并检测浓度。实验结果显示(如表1)1%sds裂解液取核酸总量约是试剂盒提取的40%,其他核酸总量较低。但由于1%sds裂解液在条件下有析出显现,因此优先选择试剂盒裂解液作为装备中裂解液。
表1.裂解液的筛选结果
b、在装置中由于存在管路较长,检测设备较多,因此需要裂解液总量较多,因此选择使用实验室配制裂解液较佳。经过上述实验证明裂解液的裂解效率不高,不足以使细胞全部裂解,从而导致提取dna总量不高,增加蛋白质变性剂的添加量,已达到高效裂解细胞的目的。选择含1%sds和10%tritonx-100的碱性裂解液分别作用于过滤了等体积海水的滤膜上和可纯培养的海洋细菌中,提取dna并测定提取浓度。实验结果显示(如表2)三种裂解液的提取效率相似,均可完成较高的细胞裂解作用。但由于sds在低温下析出,因此选择tritonx-100裂解液。
表2.裂解液对比结果
c、深海中大量存在革兰氏阳性菌,其具有较厚的细胞壁难以破坏,因此选取了可以纯培养的海洋细菌喜盐芽孢杆菌(属于厚壁菌门,革兰氏阳性菌)做补充裂解实验对比。选择含1%sds和10%tritonx-100的碱性裂解液分别作用于处于生长对数期的喜盐芽孢杆菌中,提取dna并测定提取浓度。实验结果显示(如表3)含有tritonx-100的裂解液对于该菌并没有很好的裂解作用。为此提高这两种裂解液的蛋白质变性剂的成分,选择含1%sds和20%tritonx-100的碱性裂解液重新作用于处于生长对数期的喜盐芽孢杆菌中,提取dna并测定提取浓度。实验结果显示(如表3)三种裂解液的提取效率相似,均可完成较高的细胞裂解作用。基于sds在低温下析出,因此选择tritonx-100裂解液。
表3.喜盐芽孢杆菌裂解对比结果
实施例2裂解时间
参照sds裂解液提起质粒实验中,将细菌悬浮液暴露于高ph值的碱性裂解液中,会是细胞壁破裂,染色体dna和蛋白质变性,从而将质粒dna释放到上清中。碱性溶液会是碱基配对完全破坏,但只要oh-处理的强度和时间不要太过,当ph恢复到中性时,dna双链就会再次形成。基于以上原因,设计裂解液作用时间比对实验。裂解时间选择参照试剂盒提取核酸时间作为参照,减少和延长时间,设定为1min、10min和30min作为主要时间节点,同时添加30min和1h一组对照实验。裂解液选择天根dna/rna共提试剂盒(dp422)中rlplus和配置20%tritonx-100裂解液,于过滤了2l海水的滤膜上。实验结果如下表,实验结果显示(如表4)当裂解液作用时间为30min时已经能有效的裂解海水中微生物。同时rlplus和配置20%tritonx-100裂解液的裂解效果相似,因此装备中选择含有20%tritonx-100碱性裂解液作为裂解液。
表4.裂解时间对比结果
实施例3低温实验
基于上述裂解的确定,仍然需要裂解液在深海低温条件下具有高效的裂解作用,因此设计了低温实验进行验证。配置20%tritonx-100裂解液(其他成分为tris-hcl(50mm)、nacl(150mm)、edta(1mm)、tritonx-100(20%)、蛋白酶k(20ul/ml)和溶菌酶(3mg/ml)),分成三组,一组加入1%的sds,一组加入0.05%的sds,第三组不加入sds。由于sds为强阴离子表面活性剂,能够溶解细胞膜上的脂质与蛋白,因而溶解膜蛋白而破坏细胞膜,并且解聚细胞中的核蛋白,同时sds能与蛋白质结合成为蛋白质的复合物,使蛋白质变性而沉淀。因此在低温不析出的条件下加入。使用天根dna/rna共提试剂盒(dp422)作为实验室标准提取方法作为对照。实验结果显示(如表5),在低温条件加入0.05%sds的裂解液提取dna总量约为试剂盒提取的80%,提取rna总量约为试剂盒提取的50%,符合设备要求。
表5.裂解时间对比结果
由上述实验整理得到裂解液成分如下:tris-hcl(50mm)、nacl(150mm)、edta(1mm)、tritonx-100(1%)、溶菌酶(3mg/ml)、sds(0.05%)、蛋白酶k(20μl/ml)。
实施例4吸附对比实验
在深海原位采样,核酸收集装置中关键一部分是核酸的吸附回收,基于最简、最大化原理,选取使用简单、回收效率较高的两种方式进行核酸的回收:磁珠回收和吸附柱回收。通过两种吸附方式的实验室比对实验,确定在装置设备中,采用核酸的回收方式。
实验设计
磁珠上硅羟基基团磁珠能够有效的结合核酸,从而纯化的作用,是实验室核酸纯化的最优选择之一,因此首先验证磁珠的回收效率。
对于两种方式进行核酸的回收:磁珠回收和吸附柱回收,实验设计基础是通过选用同一张滤膜,每种回收方式各一半。在其他条件相同时,分别在模拟流程中回收核酸。通过最终得到的核酸量,比较两者回收率。
实验结果如下:
a、磁珠吸附效率验证
核酸:实验室提取岸边海水dna和rna
磁珠:硅羟基基团磁珠
实验过程:使用实验室提取岸边海水的dna,取一定总量,用无菌水稀释到指定体积。首先使用注射器将磁珠注入管路中,管路中心固定在磁力架上,将dna样本使用注射器缓慢注入到管路中,流经磁力架上部磁珠所在的管路,使磁珠吸附dna。吸附完成后,回收磁珠,使用rsb(resuspensionbuffer)回收dna,测定回收后dna浓度并计算会后效率。rna吸附实验同上。实验结果显示(如表6)磁珠在管路中均聚集在磁力架边缘,无法有效、均匀的混合于dna样品,导致吸附回收的dna总量不高。dna回收效率随吸附体积的增大而减小,回收效率不足10%;rna回收效率随吸附体积的增大而减小,当体积增加时,甚至回收不到。因此不选用磁珠吸附回收方式。
表6.磁珠吸附效率对比结果
b.回收效率对比验证
样品:2l海水
磁珠:硅羟基基团磁珠
吸附柱:天根试剂盒吸附柱cb5(tiangendp332)
针对磁珠回收和吸附柱回收,同时采用等量2l的海水进行磁珠吸附和吸附柱吸附的效率对比实验。首先使用47mm直径的0.2um滤膜过滤2l海水,35mlsds裂解液冲洗滤膜,回收裂解液。重复两组。然后一组裂解液加入等体积的无水乙醇,用蠕动泵将混合液压过吸附柱cb5(tiangendp332)吸附dna。200ultebuffer回收dna。最后另一组回收到的裂解液约为15ml,使用100ul硅羟基基团磁珠进行dna回收。实验结果显示(如表7)磁珠吸附的dna总量(17.5ng)约是吸附柱吸附dna总量(488ng)的3.6%,因此选择吸附柱作为核酸收集管。
表7.回收方式对比结果
4、吸附条件对比实验
对于设备中选用的吸附柱,基于最简化原则,测试裂解后溶液中是否必须加入乙醇溶液,才能有效吸附核酸。
实验设计
为保证实验存在准确的对比性,实验使用同一张滤膜,在其他条件相同仅有乙醇加入的不同,测定提取到的核酸浓度。实验流程参照图2。
实验结果如下:
核酸提取试剂盒:天根dna/rna共提试剂盒(dp422)
滤膜:0.2um滤膜(milliporetm,直径47mm)
处理方法:①滤膜首先使用3mg/ml溶菌酶处理10min;②试剂盒裂解液处理10min;③吸取混合液后将液体分成两部分,一部分加入乙醇,另一部分不加入乙醇,使用试剂盒中吸附柱提取核酸。重复两组实验。
实验结果:从测定rna浓度可以明显看出当混合液体中没有乙醇溶液时,吸附柱不能够选择吸附上rna(浓度测定值如表8)。
表8.吸附条件对比结果
实施例5替代效果检测
在深海原位采样,核酸收集装置中,存在管路切换、管路内液体相互转换过程。针对两种液体之间相互转换并发生替代,直至完全转换的过程,设计管路替换实验。根据实验数据,记录裂解液和保存液的初始需要携带体积,保证裂解液能够充分作用于富集腔内,完成裂解工作,得到总量更多、质量更高的核酸,从而获得更多关于深海原位环境的信息。
因此设计使用管路联合方式模拟装置,从最初纯水注入开始,到检测海水注入、裂解液注入和海水填充的过程,完成整体流程。测定回收到的核酸量是否达到考核标准。
实验设计
设计使用管路联合方式模拟装置。根据原位裂解实验流程,存在溶质含量较低、均匀液体之间替换过程和高盐度和低盐度液体之间替换甚至混合过程。因此设计如下实验:
(1)均匀液体混合,测定吸光度
对于溶质含量较低、均匀液体之间替换过程,设计使用吸光度在300nm处存在最大吸光值的带颜色溶液和纯水,作为两种替换溶液。当吸光度值的范围从0变成替换后溶液的吸光度值时,即可认定为管路内发生了完全替换。实验装置使用实验室过滤器,可装载142mm滤膜过滤器。
(2)盐溶液混合,测定液体中盐浓度
对于较高盐浓度的液体和纯水之间的替换实验,设计使用配制盐溶液0.25m,即盐浓度约为15.4ppt。当盐浓度范围从0ppt变成配制盐溶液浓度,即可认定为管路内发生了完全替换。实验装置使用实验室过滤器,可装载142mm滤膜过滤器。
(3)盐溶液混合,测定液体中盐浓度
上述实验使用实验室过滤器,需要进一步确认使用富集装置在管路替换过程中的替换效率。实验使用配制氯化钠溶液(浓度参照已经在深海采样中使用的rna样品保存液浓度的1/2,配制氯化钠溶液浓度为24.7ppt,该浓度为稀释1/10的浓度,在检测设备测定范围内。)和已经经过过滤的海水进行替换实验。当盐浓度范围从0.3ppt变成配制盐溶液浓度,即可认定为管路内发生了完全替换。实验装置使用原位富集装置,可装载142mm滤膜。
实验结果如下:
(1)均匀液体混合,测定吸光度
首先在整个管路和过滤器中加入纯水填充,确保管路内没有气泡(设备内管路及各部分元件,都需要有液体填充,在深海中不能有气泡存在),实验开始时,在过滤器底端收集混合后液体,每15ml为一个单位,定为一个测量点。分别收集25、40和40个测量点,分别测定每个测量点在300nm处的吸光度值。实验重复进行三次。
设备:实验室用过滤器
均匀液体:纯水、替换溶液(1.8l水+200ml饮料)
初始吸光度:设置纯水的吸光度值为0,替换溶液吸光度为0.83
设备+管路内体积:300ml
实验过程:管路提前注满纯水,注满体积约为300ml。第一次注入替换溶液开始,丢弃200ml水(由于管路内体积较大,因此丢弃前半部分管路内纯水),然后开始收集,每次15ml,使用分光光度计检测吸光度值(每个测量点均做三次测定后取平均值)。第二、三次从开始注入替换溶液开始同时收集。实验结果如下图3、4、5所示。对于均匀液体混合,管路中完全替换的体积约是整体管路内容积。参照上述试验约为300ml。
(2)盐溶液混合,测定液体中盐浓度
首先在整个管路和过滤器中加入纯水填充,确保管路内没有气泡,实验开始时,在过滤器底端收集混合后液体,每15ml为一个单位,定为一个测量点。收集50个测量点。实验重复进行两次。
设备:实验室用过滤器
均匀液体:纯水、替换溶液(0.25m氯化钠溶液,该浓度为随机选择,浓度在盐度计检测范围内)
初始盐浓度:检测纯水的盐浓度为0,替换溶液盐浓度为15.4ppt
设备+管路内体积:300ml
实验过程:管路提前注满纯水,注满体积约为300ml。两次实验均从开始开始注入替换溶液即开始收集,每次15ml。实验结果如图6、7所示。对于不同浓度盐溶液混合,管路中完全替换的体积约是整体管路内容积的两倍。参照上述试验约为600ml。
(3)盐溶液混合,测定液体中盐浓度
首先在整个管路和过滤器中加入纯水填充,确保管路内没有气泡,实验开始时,在过滤器底端收集混合后液体,每约12ml为一个单位,定为一个测量点。收集50个测量点。实验重复进行三次。
设备:原位富集装置
均匀液体:使用0.22um滤膜过滤后的海水、替换溶液(3.4m氯化钠溶液,该浓度为常用保存rna样品的保存试剂rnalater浓度的1/2)
初始盐浓度:检测海水的盐浓度为0.35ppt,替换溶液盐浓度为24.7ppt(原溶液稀释1/10后测定)
设备+管路内体积:165ml
实验过程:管路提前注满过滤后海水,注满体积约为165ml。每次实验均从开始开始注入替换溶液即开始收集,每次15ml。实验结果如图8、9、10所示。替换约在第9个点即可替换80%,体积约是120ml,占总体积的70%左右(总体积包括滤腔内体积约50ml和富集装置内体积115ml)。工作中预设替换体积应与总内体积相等。
实施例6核酸保存实验
富集在生物滤膜腔的微生物,经过裂解后,释放出核酸,同裂解液经由管路到达核酸收集管处,被吸附柱所选择性吸附。在这过程中,核酸一直存在于裂解液中,收到裂解液的保护作用,使得核酸不会发生降解。当核酸被选择性吸附后,与吸附柱结合后,仍然需要保护液的保护,已达到在水下长时间作业的情况下,不会发生降解。因此需要选择保护液对核酸进行保护作用。
实验设计
将吸附在吸附柱上的核酸保存在裂解液和乙醇的混合液中,低温保存过夜,洗脱核酸,检测核酸提取量是否发生变化,计算核酸降解量是否在可接受范围内。
由于模拟设备在深海工作环境,吸附柱结束吸附工作后,由核酸裂解液及酒精的混合液作为保护液,本实验吸附柱核酸虽有降解,但过滤时有保护液中的核酸作为补给,降解和损失的速度是缓慢的。(图11)
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!