一种基于动态共价键的高分子网络拓扑异构体系及其应用方法与流程
本发明属于新型功能材料领域,具体涉及一种基于动态共价键的高分子网络拓扑异构体系及其应用方法。
背景技术:
高分子动态共价网络是一类网络内部含有动态共价键的聚合物材料。在外界刺激(包括加热、光照、氧化还原等)的作用下,网络中的动态共价键会不断地发生键的断裂与重组。
传统的高分子动态共价网络主要应用于以下领域:1)聚合物材料的自修复:聚合物材料在遭到物理损伤后,破损处(裂缝或缺口等)的聚合物网络也随之被破环。通过激活网络内部动态共价键的键交换反应,网络可以发生动态的断裂和重组,因此,被破坏的网络可以重新连接成原始的完整网络结构,从而实现材料的自修复(cheng,nat.commun.,2014,5,3218);2)形状记忆材料复杂原始形状的构建:形状记忆材料是一类可程序化变形的智能材料,可在外界刺激下从临时形状回复到永久形状。在施加应力使形状记忆材料发生变形后,通过激活网络内部动态共价键的键交换反应,使得加载于材料的应力发生松弛,变形后的临时形状就被固定成形状记忆聚合物的原始形状(xie,sci.adv.2016,2,e1501297);3)聚合物材料的重加工:通常的热固性聚合物由于其交联网络的特性不具备重加工的能力,但是在高分子网络中引入动态共价键以后,与改变形状记忆材料永久形状相类似,被激活的动态共价键会通过应力松弛,固定住材料二次模塑后的形状,实现聚合物材料的重加工(xie,acsappl.mater.interfaces,2017,9,22077.)。因此,高分子动态共价网络已经成为聚合物材料领域中的一个研究热点。
尽管如此,上述所提及的高分子动态共价网络的拓扑结构在激活动态共价键前后并未发生变化。因为传统的高分子动态共价网络的拓扑结构是处于热力学稳定的状态,例如其各个区域化学势,网链构象数都处于热力学稳定的状态。对于高分子材料而言,材料的物性例如结晶度,相转变温度和模量等都与高分子网络的拓扑结构直接相关。因此,传统的高分子动态共价网络难以实现高分子相关物性的改变和调控。
技术实现要素:
本发明提供了一种基于动态共价键的高分子网络拓扑异构体系及其应用方法,本发明提供的高分子网络拓扑异构体系中的拓扑结构可以根据需要发生特定的异构,实现对聚合物相关物性的调控,并且拓扑结构的异构过程可以区域化,分步多次地进行。
本发明提供如下技术方案:
一种基于动态共价键的高分子拓扑异构网络体系,所述高分子拓扑异构网络体系为含有动态共价键的交联型聚合物,所述高分子拓扑异构网络体系中的拓扑结构处于由动态共价键暂时固定的热力学非稳态;在键交换催化剂的作用和温度达到动态共价键激活温度以上时,所述动态共价键发生键交换反应和拓扑结构发生改变。
所述高分子拓扑异构网络体系可以通过含有动态共价键的聚合物前驱体通过一定的交联化学制备,或者由两种及两种以上不含动态共价键的单体通过化学反应生成动态共价键并形成交联网络而制备。
在发明中,热力学非稳态可以是由网络区域化学势或网链构象数处于非稳态中的一种或多种因素所造成的。在动态共价键未被激活时,材料(聚合物或高分子拓扑异构网络体系)的网络结构由于共价键的固定作用临时处于动力学的稳态,材料的相关物性也因此保持稳定。但是当温度达到动态共价键激活温度以上时,高分子网络的拓扑结构会在熵的驱动下,通过动态共价键的键交换反应逐渐向热力学更稳定的方向演化,例如向网络中区域化学势降低,网链构象数增多的拓扑结构改变。这样的网络拓扑结构的变化称为高分子网络的拓扑异构过程。由于动态共价键的交换速率可以通过催化剂的含量进行调节,因此,在相同的键交换反应时间内,高分子网络的拓扑异构程度可以由催化剂的相对含量进行调控。对于高分子材料而言,其众多物性例如结晶度、模量和相转变温度都与其网络的拓扑结构直接相关。因此,通过控制上述的高分子网络拓扑异构程度就可以实现对高分子材料相关物性的调控。
所述动态共价键包括但不限于酯键、氨酯键、脲键、二酮氨基键、硅氧键或碳碳双键中的一种或多种。
在本发明中,所述动态共价键适用于任意一种高分子拓扑异构网络体系。
所述聚合物体系的动态共价键激活温度可以通过相应催化剂体系来调节,根据应用设定为20-150℃或者更宽的范围。为保证聚合物网络的拓扑结构在室温下保持稳定(被未激活的动态共价键临时固定为动力学稳态),作为优选,所述动态共价键激活温度为60-150℃。
作为优选,本发明以特定动态共价键为例,对上述两种不同类型的高分子拓扑异构体系的设计原则分别进行说明。
当所述的网络热力学非稳态结构是由网络区域化学势过高所造成时,所述高分子拓扑异构网络体系的网络结构包括至少两种不同化学组成的嵌段网链结构,所述的嵌段网链结构之间包含可以相互反应的动态共价键(简称为网络体系ⅰ)。当嵌段网链结构之间的动态共价键在键交换催化剂和合适温度下发生键交换反应时,高分子网络会从嵌段型的网络结构(网络区域化学势相对较高)向混合型网络结构(网络区域化学势相对较低)演化为一种化学势驱动的网络拓扑异构。
对于网络体系ⅰ,当所述动态共价键为酯键时,所述的含有动态共价键的聚合物网络(高分子拓扑异构网络体系)可以由两种及两种以上含酯基的聚合物前驱体直接交联而成,也可以利用两种及两种以上不含酯键的单体反应生成酯键并交联而成,含有酯键的聚合物前驱体可以选自聚酯多元醇,含端双键的饱和/不饱和聚酯等;不含酯键的单体可以选自环氧基化合物。
作为优选,聚酯多元醇可以选自聚己内酯二醇,聚十五内酯二醇,聚己二酸乙二醇酯二醇,聚己二酸乙二醇-丙二醇酯二醇,聚己二酸一缩二乙二醇酯二醇中的两种或多种。此时,交联剂可以选自能与多元醇反应的多元酸、多元异氰酸酯等。作为进一步优选,交联剂可以使用多元异氰酸酯,包括但不限于以下化合物:六亚甲基二异氰酸酯,三苯甲烷三异氰酸酯,hdi三聚体等。
作为优选,含端双键的饱和/不饱和聚酯可以选自聚己内酯二(甲基)丙烯酸酯,聚十五内酯二(甲基)丙烯酸酯,聚丙交酯二(甲基)丙烯酸酯,聚乙交酯二(甲基)丙烯酸酯,聚邻苯型和间苯型不饱和二(甲基)丙烯酸酯中的两种或多种。此时,交联剂可以使用多巯基化合物,包括但不限于以下化合物:四(3-巯基丙酸)季戊四醇酯,季戊四醇四巯基乙酸酯,三羟甲基丙烷三(3-巯基丙酸酯)等。
作为优选,环氧基化合物可以选自酚醛环氧树脂,双酚a二环氧甘油醚,双酚f二环氧甘油醚,季戊四醇四环氧甘油醚,四溴双酚a二环氧甘油醚,1,6-己二醇二环氧甘油醚,双酚a聚乙二醇环氧甘油醚,环氧化多不饱和脂肪酸,环氧化植物油等。此时,可以选择合适的多元酸或酸酐来与上述环氧化合物进行开环反应生成酯基,作为聚合物的交联位点。作为优选,多元酸可以选自包含2-40个碳原子的脂肪族二酸(戊二酸,己二酸,庚二酸,辛二酸,壬二酸,葵二酸,十二烷二酸等)及其混合物,不饱和脂肪酸(十一碳烯酸,棕榈油酸,油酸,亚油酸,亚麻酸,蓖麻油酸,二十二碳烯酸等)的低聚物,也可以选自含2-40个碳原子的芳香族二酸(如邻苯二酸,偏苯三酸,对苯二酸或萘二甲酸等)及其混合物;作为优选,酸酐可以选自邻苯二酸酐,甲基迪克酸酐,六氢邻苯二酸酐,十二碳烯基丁二酸酐,五二酸酐等。
对于网络体系ⅰ,当所述动态共价键为氨酯键时,所述的含有氨酯键的聚合物网络可以由两种及两种以上多元醇和多元异氰酸酯进行聚合反应交联制备得到。
作为优选,所述的多元醇为聚酯多元醇,聚醚多元醇和2-40个碳原子的小分子多元醇中的一种或多种,进一步优选,所述的聚酯多元醇为聚己内酯二醇,聚己二酸乙二醇酯二醇,据己二酸乙二醇-丙二醇酯二醇,聚己二酸一缩二乙二醇酯二醇,聚己二酸-1,4-丁二醇酯二醇,聚己二酸乙二醇-1,4-丁二醇酯二醇中的一种或多种。所述的聚醚多元醇优选为聚醚多二元醇,进一步优选为聚氧化乙烯二醇,聚四氢呋喃二醇,四氢呋喃-氧化丙烯共聚二醇中的一种或多种。此时,所述的多元异氰酸酯选自聚六亚甲基二异氰酸酯,二苯甲基二异氰酸酯,三苯甲烷三异氰酸酯和六亚甲基异氰酸酯的三聚体中的一种或多种。
对于网络体系ⅰ,当所述动态共价键为脲键时,所述的含有脲键的聚合物网络可以由两种及两种以上多元胺和多元异氰酸酯进行聚合反应交联制备得到。
作为优选,所述的多元胺为2-40个碳原子的线性二元伯胺和线性二元仲胺中的一种或多种,进一步优选,所述线性二元伯胺为二叔丁基乙二胺,二叔丁基丁二胺中的一种或多种。此时,所述的多元异氰酸酯选自聚六亚甲基二异氰酸酯,二苯甲基二异氰酸酯,三苯甲烷三异氰酸酯和六亚甲基异氰酸酯的三聚体中的一种或多种。
对于网络体系ⅰ,当所述动态共价键为碳碳双键时,所述的含有碳碳双键的聚合物网络可以由两种及两种以上不饱和大分子前驱体通过交联制备,也可以通过两种及两种以上环烯烃单体通过开环易位形成前驱体后交联而成。
作为优选,所述的不饱和大分子前驱体可以选自聚丁二烯,聚异戊二烯,聚降冰片烯,聚乙叉降冰片烯等,此时,聚合物网络可以通过加入自由基引发剂进行交联制备,包括但不限于偶氮二异丁腈,过氧化苯甲酮,过氧化氢异丙苯,过氧化二叔丁基,4-羟基二苯甲酮,9,10-菲二酮,2-乙基蒽醌等。聚合物网络也可以通过加入硫醇交联剂利用machel-addition反应制备,此时,硫醇交联剂可以使用多巯基化合物,包括但不限于以下化合物:四(3-巯基丙酸)季戊四醇酯,季戊四醇四巯基乙酸酯,三羟甲基丙烷三(3-巯基丙酸酯)等。
作为优选,所述的环烯烃单体可以选自5-降冰烯-2-基乙酸酯,5-降冰片烯-2-羧酸,5-降冰片烯-2-甲腈,5-降冰片烯-2,3-二甲醇,5-降冰片烯-2-甲胺,二环[2.2.1]庚-5-烯-2-甲醇,5-降冰片烯-2-羧酸甲酯,5-降冰片烯-2,3-二甲酰亚胺和羟基二环戊二烯等衍生物中的两种及以上。当所选取的单体开环易位后的前驱体除碳碳双键无其他特征官能团时,聚合物网络可以通过加入自由基引发剂进行交联制备,包括但不限于偶氮二异丁腈,过氧化苯甲酮,过氧化氢异丙苯,过氧化二叔丁基,4-羟基二苯甲酮,9,10-菲二酮,2-乙基蒽醌等。聚合物网络也可以通过加入硫醇交联剂利用machel-addition反应制备,此时,硫醇交联剂可以使用多巯基化合物,包括但不限于以下化合物:四(3-巯基丙酸)季戊四醇酯,季戊四醇四巯基乙酸酯,三羟甲基丙烷三(3-巯基丙酸酯)等。当所制备的前驱体含有其他特征官能基团时(例如羟基,羰基,胺基等),作为优选,当特征官能基团为羟基或胺基时,交联剂可以使用多元异氰酸酯,包括但不限于以下化合物:六亚甲基二异氰酸酯,三苯甲烷三异氰酸酯,六亚甲基二异氰酸酯三聚体等。当特征官能基团为羰基时,交联剂可以选自多元胺,包括但不限于2,2'-氧基二乙胺,1,5-戊二胺,二叔丁基乙二胺,二叔丁基丁二胺等。
对于网络体系ⅰ,当所述动态共价键为二酮氨基键时,所述的含有二酮氨基键的聚合物网络可以由两种及两种以上二元胺和双乙酰乙酸化合物缩聚形成大分子前驱体并交联而成。作为优选,二元胺可以选自2-40个碳原子的线性二元伯胺或仲胺,进一步优选,包括但不限于2,2'-氧基二乙胺,1,5-戊二胺,二叔丁基乙二胺,二叔丁基丁二胺,对二亚基甲苯二胺,间二亚甲基苯二胺,对二亚乙基苯二胺,间二亚甲基苯二胺等。此时交联剂可以使用多元胺,包括但不限于以下化合物:三(2-氨基乙基)胺,1,5,9-三氮杂环十二烷等。
当所述的网络热力学非稳态结构由网链构象数过低所造成时,主要可以分为以下两种:1)重新分布聚合物网络中的网链链长,在动态共价键被激活时,具有不同链长的分子链(网络构象数相对较少)之间会通过键交换反应转变为链长相对均一的分子链(网络构象数相对较多),高分子网络的拓扑异构过程通常为由接枝型网络向梳状型网络演化;2)改变网链结构,在动态共价键被激活时,含有动态共价键的位点之间发生交换,使得网络分子链发生转移,断裂等,网络结构中分子链构象数增加。
对于所述异构化过程为重新分布链长的高分子网络拓扑异构体系而言,所述高分子拓扑异构网络体系的网络结构主要包括网络主链框架,接枝动态长侧链和悬挂短链(相对于接枝长链而言)侧基三个部分(简称为网络体系ⅱ)。为保证高分子网络拓扑异构行为更加可控,作为优选,网络主链框架选自通常不能发生动态共价键交换反应的碳碳单键、碳氧单键或碳硫单键中的一种或多种,而接枝动态长链和悬挂短链侧基则要求相互之间可以发生键交换反应。
对于网络体系ⅱ,当所述动态共价键为酯键时,含有动态共价键的网络中的接枝动态长链和悬挂短链侧基之间会发生酯交换反应,作为优选,悬挂短链侧基可以选自单端双键的小分子醇或小分子酯,接枝动态长链可以选自单端双键的饱和聚酯,进一步优选,悬挂短链侧基可以选自含有单个碳碳双键的一元醇或多元醇、含有多个碳碳双键的一元醇或多元醇,接枝动态长链则可以选上述由悬挂短链侧基单体引发的各类饱和聚酯的一种或多种。
作为优选,含有单个碳碳双键的一元醇或多元醇可以选自2-(2-乙烯氧基乙氧基)乙醇,2-乙烯氧基乙醇,4-乙烯氧基丁醇,2-烯丙氧基乙醇,3-烯丙氧基-1,2-丙二醇中的一种或多种,长侧链可以选自由上述短侧链单体引发的聚乙交酯,聚丙交酯,聚己内酯,聚十五内酯的一种或多种。此时交联剂可以选自能与双键反应的多官能度交联剂,包括但不限于以下化合物:对二乙烯基苯,二乙二醇二乙烯醚,四烯丙氧基乙烷,三聚氰酸三烯丙酯等。
作为优选,含有多个碳碳双键的一元醇或多元醇可以选自丙三醇α,α'-二烯丙基醚,1,5-己二烯-3,4-二醇,2,2-二[(2-丙烯基氧)甲基]-1-丁醇中的一种或多种,长侧链可以选自由上述短侧链单体引发的聚乙交酯,聚丙交酯,聚己内酯,聚十五内酯的一种或多种。此时网络体系需要二硫醇作为扩链剂形成主链结构,包括但不限于以下化合物:1,3-丙二硫醇,1,4-丁二硫醇,1,6-己二硫醇,3,6-二氧杂-1,8-辛烷二硫醇等。此时交联剂为多官能度的双键交联剂,包括但不限于以下化合物:对二乙烯基苯,二乙二醇二乙烯醚,四烯丙氧基乙烷,三聚氰酸三烯丙酯等。
对于网络体系ⅱ,当所述动态共价键为硅氧键时,含有动态共价键的网络可以由两种及两种以上含有活性反应基团的有机硅化合物通过一定的反应交联而成。作为优选,反应活性基团可以选自硅氧键,乙烯基,巯基,氨基,环氧基,羧基,(甲基)丙烯酸酯,烷氧基等。进一步说明,当活性基团为硅氧键或乙烯基时,聚合物网络可由含硅氧键的有机硅化合物与含有乙烯基有机硅化合物在金属铂催化剂下进行硅氢加成反应得到;当活性基团为巯基时,聚合物网络可由含巯基有机硅化合物与含有乙烯基或(甲基)丙烯酸酯基有机硅化合物发生thiol-ene反应或者麦克尔加成反应得到;当活性基团为含(甲基)丙烯酸酯基时,聚合物网络可由含(甲基)丙烯酸酯基有机硅化合物通过自由基反应得到;当活性基团为环氧基时,聚合物网络可由含环氧基有机硅化合物与氨基(或羧基,酸酐)有机硅化合物反应得到。
对于所述异构化过程为改变网链结构的高分子网络拓扑异构体系而言,其网络结构包括具有键交换位点的聚合物主链和小分子侧基两个部分((简称为网络体系ⅲ))。小分子侧基与聚合物主链之间会发生键交换反应,从而聚合物主链在网络结构中发生转移,断裂等行为,造成网链结构的变化。
对于网络体系ⅲ,当所述动态共价键为酯键时,聚合物主链可以选自双端(甲基)丙烯酸酯双键修饰的大分子前驱体,作为优选,可以选自聚己内酯二(甲基)丙烯酸酯,聚十五内酯二(甲基)丙烯酸酯,聚丙交酯二(甲基)丙烯酸酯,聚乙交酯二(甲基)丙烯酸酯,聚乙二醇二(甲基)丙烯酸酯中的一种或多种。小分子侧基可以选自含端羟基的(甲基)丙烯酸衍生物,作为优选,可以选自羟甲基(甲基)丙烯酸酯,羟乙基(甲基)丙烯酸酯,羟丙基(甲基)丙烯酸酯,羟甲基(甲基)丙烯酰胺,羟乙基(甲基)丙烯酰胺,羟丙基(甲基)丙烯酰胺中的一种或多种。
上述高分子拓扑异构网络体系在应用过程中还需要加入动态共价键键交换催化剂。所述的键交换催化剂可以在网络合成过程中直接引入,也可以在材料应用时通过光潜酸或光潜碱催化剂光照引入。
当所述键交换催化剂为高分子拓扑异构网络合成过程中直接引入时,催化剂可以选自1,5,7-三氮杂二环[4.4.0]葵-5-烯、有机伯胺,锌盐、锡盐、镁盐、钴盐、钙盐,grubs第一代催化剂,grubs第二代催化剂等中的一种或多种。
当所述催化剂为材料应用时的光潜酸或光潜碱催化剂时,催化剂为1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物,双(环己磺酰基)重氮甲烷,(4-甲基苯基)二苯基锍三氟甲烷基磺酸等光产酸或光产碱中的一种或多种。进一步优选,为了便于在材料不同区域引入所需的不同含量的催化剂,催化剂采用1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物;该催化剂可以在紫外光照的刺激下释放出键交换催化剂(1,5,7-三氮杂二环[4.4.0]葵-5-烯),并且该过程中释放出的催化剂含量与光照时间成正比。因此,该体系可以通过控制光照时间来实现对材料内部的键交换催化剂浓度的控制。键交换催化剂在聚合物体系中所占的质量分数为至少0.5%。
本发明提供的高分子拓扑异构网络的应用方法包括如下步骤:
当所述键交换催化剂为高分子拓扑异构网络合成过程中直接引入时,其应用操作步骤如下:
1)当温度加热到动态共价激活温度以上时,具有原始网络拓扑结构i的聚合物发生进行键交换反应,高分子网络的拓扑结构从而发生改变,得到网络拓扑结构ii;
2)冷却,步骤1)中的高分子网络拓扑结构被固定,成为聚合物的一个新的网络拓扑结构ii,材料的相关物性发生改变。
当所述催化剂为材料应用时的光潜酸/碱催化剂时,其应用操作步骤如下:
1)当温度加热到动态共价键激活温度以上时,照射材料的特定区域,通过控制光照条件控制材料内部的催化剂浓度分布;
2)保持温度,具有原始网络拓扑结构i的聚合物发生进行键交换反应,高分子网络的拓扑结构从而发生改变,得到网络拓扑结构ii;
3)冷却,步骤2)中的高分子网络拓扑结构被固定,成为聚合物的一个新的网络拓扑结构ii,材料的相关物性发生改变。
其中,在高分子拓扑异构网络达到热力学稳态之前,拓扑异构过程可以多次在聚合物材料使用过程中的任意阶段、多次叠加地进行。其中,材料相关物性包括结晶度、相转变温度、模量中的一种或多种。所述的相转变温度为聚合物的玻璃化温度或熔融温度。
与现有技术相比,本发明的有益效果体现于:
本发明提出的基于动态共价键的高分子拓扑异构网络体系通过特定的分子设计,使高分子网络拓扑结构在合成时处于热力学非稳态,并在熵的驱动下借助动态共价键键交换反应使聚合物网络的拓扑结构从热力学非稳态向热力学稳态演化,从而完成高分子网络的拓扑异构的过程,并成功地实现对材料与网络拓扑结构相关的物性的连续调控。
本发明可以紫外光照的方法在材料特定区域引入特定浓度的催化剂,从而实现在单一材料内部引入不同的高分子网络拓扑异构程度,实现了聚合物材料特定区域特定物性的调控,为设计非均质连续材料提供了新思路。
附图说明
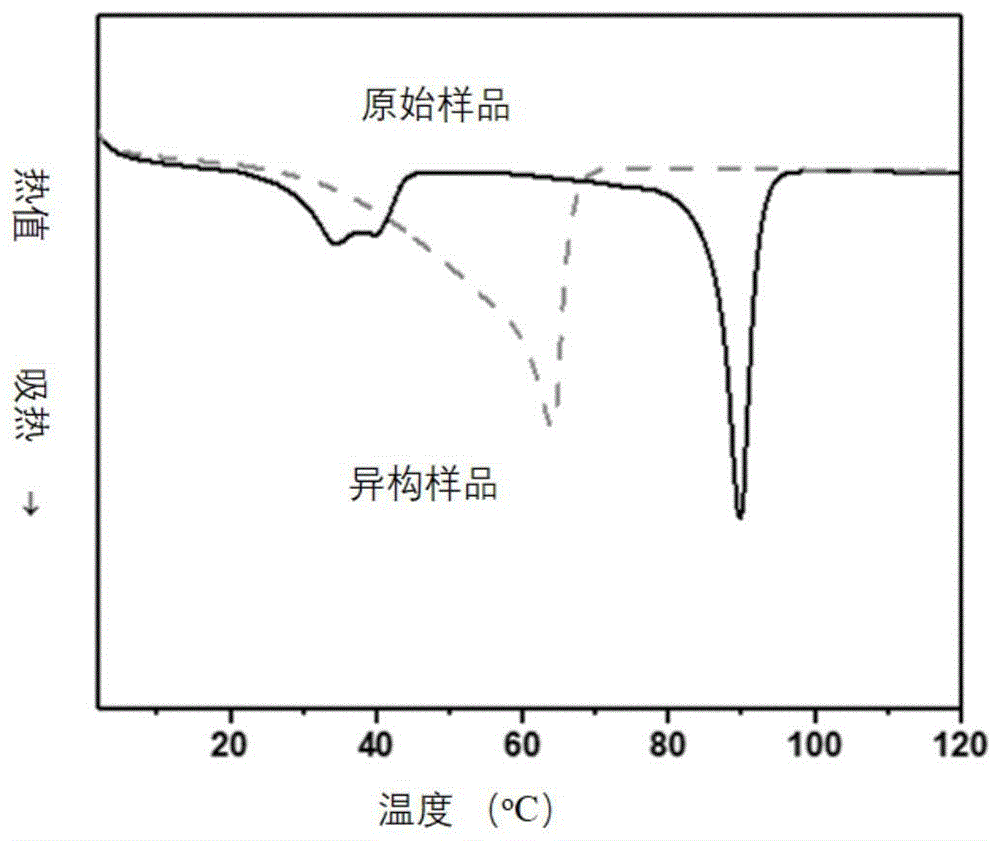
图1为实施例1的高分子网络拓扑异构前后的熔点变化图;
图2为实施例1的高分子网络拓扑异构前后的应力应变曲线;
图3为实施例2在不同光照时间下的熔点变化图;
图4为实施例2在不同光照时间下的结晶度和模量变化图;
图5为实施例2在不同光照时间下的应力应变曲线图;
图6为实施例3在不同光照时间下的熔点变化图;
图7为实施例3在不同光照时间下的结晶度变化图;
图8为实施例4在网络异构前后玻璃化转变温度变化图;
图9为实施例4在网络异构前后应力应变曲线图;
图10为实施例5在网络异构前后玻璃化转变温度变化图;
图11为实施例2材料不同区域物性调控的具体过程;
图12为化学势驱动的高分子网络拓扑异构示意图;
图13为网链构象数驱动的高分子网络拓扑异构-重新分配链长型示意图;
图14为网链构象数驱动的高分子网络拓扑异构-改变网链结构型意图。
具体实施方式
以下结合实施例对本发明做进一步说明,但本发明要求的保护的范围并不局限于实施例表达的范围。
实施例1
原料:
a)聚己内酯二醇(pclda):mw=10000,sigma-aldrich公司;结构式如下:
b)聚十五内酯二醇(ppdlda):mw=2000,sigma-aldrich公司;
结构式如下:
c)六亚甲基二异氰酸酯(hdi):sigma-aldrich公司;结构式如下:
d)二月桂酸二丁基锡(dbtdl):tci公司;
e)1,5,7-三氮杂二环[4.4.0]葵-5-烯:tci公司;
f)二氯乙烷:阿拉丁试剂(上海)有限公司;
制备方法:
称取0.1mmol的聚十五内酯二丙烯酸酯,0.9mmol的聚己内酯二丙烯酸酯酯和1mmol的六亚甲基二异氰酸酯在10ml二氯乙烷中(其中pcl-diol和ppdl-diol的总羟基数与异氰酸酯基团的摩尔比为1:1),加热至80c溶解。随后加入二月桂酸二丁基锡(其加入量为体系总质量的0.5%),1,5,7-三氮杂二环[4.4.0]葵-5-烯(其加入量为体系总质量的1%)搅拌使其完全溶解。将上述溶液倒入密封玻璃槽中,置于80°烘箱反应12小时。将固化后的样品取出,置于真空干燥箱内,80℃干燥24小时。
高分子网络拓扑结构异构过程:将样条加热至120摄氏度,保持不同的反应时间。在冷却后,对材料的相关物性进行测试。例如:dsc测试材料熔点的变化,利用拉伸机(80℃条件下)测试模量和断裂伸长率的变化;及测量玻璃化转变温度和利用afm测试材料的相分离程度。
实施例2
原料:
a)2,2-二[(2-丙烯基氧)甲基]-1-丁醇:tci公司;结构式如下:
b)己内酯:tci公司;结构式如下:
c)3,6-二氧杂-1,8-辛烷二硫醇,tci公司;其结构式如下:
d)四烯丙氧基乙烷,tci公司;其结构式为:
e)1-羟基环己基苯基甲酮(uv-184):tci公司;
f)辛酸亚锡:tci公司;
g)1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物:tci公司;
h)甲苯:阿拉丁(上海)试剂公司;
i)正己烷:阿拉丁(上海)试剂公司;
制备方法:
称取0.35mol的己内酯,8mmol的2,2-二[(2-丙烯基氧)甲基]-1-丁醇和0.5mmol辛酸亚锡,在氩气氛围下加热至120℃反应10小时。将所得到的产物溶解于50ml的甲苯后,再用正己烷沉淀得到白色粉状的2,2-二[(2-丙烯基氧)甲基]-1-丁醇-聚己内酯(分子量为5000da),产物置于于真空干燥箱内,80c干燥24小时。随后称取0.7mol的2,2-二[(2-丙烯基氧)甲基]-1-丁醇-聚己内酯,1mol的2,2-二[(2-丙烯基氧)甲基]-1-丁醇和0.126mol3,6-二氧杂-1,8-辛烷二硫醇于10ml的甲苯中,加热至80℃溶解。随后加入1-羟基环己基苯基甲酮(其加入量为体系总质量的0.5%),搅拌均匀后置于紫外灯(紫外波长为365nm)下照射5分钟,随后加入0.09mol的四烯丙氧基乙烷和1-羟基环己基苯基甲酮(其加入量为体系总质量的0.5%),搅拌使其完全溶解。将上述溶液倒入密封玻璃槽中,至于紫外灯(紫外波长365nm)下照射5分钟。将固化后的样品取出,至于真空干燥箱内,80℃干燥24小时。随后将样品浸泡在1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物的甲苯溶液中(其络合物的质量分数为1.5%)约1小时,使其充分溶胀,随后置于真空干燥箱内,80℃干燥24小时。
高分子网络拓扑结构异构过程:将样条加热至80摄氏度,随后将材料置于紫外灯(紫外灯波长为365nm)下,照射特定的时间,促使1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物释放酯交换催化剂,随后保持80℃约2小时。在冷却后,对材料的相关物性进行测试。例如:利用dsc测试材料熔点和结晶度的变化;利用拉伸机测试模量和断裂伸长率的变化,利用afm测试材料的相分离程度。
实施例3
原料:
a)聚乙二醇丙烯酸酯(pegda):分子量mn=3458,sigma-aldrich公司;结构式入下:
b)羟乙基丙烯酰胺:sigma-aldrich公司;结构式如下:
c)过氧化苯甲酮:sigma-aldrich公司;
d)1,5,7-三氮杂二环[4.4.0]葵-5-烯:tci公司;
e)n,n-二甲基甲酰胺(dmf):tci公司;
制备方法:
称取0.5gpegda和0.07g羟乙基丙烯酰胺在0.5mldmf中,在60℃下形成澄清溶液,随后加入bpo(其加入量为体系质量的1.5%)和1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物(其加入量为体系质量的1.5%)。随后将前驱体溶液转移至密封玻璃槽中,保持100℃反应24小时。将所得到的薄膜与在80℃真空烘箱中干燥24小时。
高分子网络拓扑结构异构过程:将样条加热至80摄氏度,随后将材料置于紫外灯(紫外灯波长为365nm)下,照射特定的时间,促使1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物释放酯交换催化剂,随后保持80℃约2小时。在冷却后,对材料的相关物性进行测试。例如:利用dsc测试材料熔点和结晶度的变化;利用拉伸机测试模量和断裂伸长率的变化,利用afm测试材料的相分离程度。
实施例4
原料:
a)单氨基封端聚(二甲基硅氧烷),分子量分别为1000,5000:百灵威公司;结构式如下:
b)异氰酸酯丙烯酸乙酯:tci公司;其结构式如下:
c)六亚甲基二异氰酸酯:tci公司;其结构式如下:
d)4-氰基-4-[(十二烷基硫烷基硫羰基)硫烷基]戊酸:tci公司;
e)偶氮二异丁腈(aibn):tci公司;
f)1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物:tci公司;
g)二甲苯:阿拉丁(上海)试剂公司;
制备方法:
分别称取0.5mol和0.2mol分子量为1000和5000的单氨基封端聚(二甲基硅氧烷)溶解于5ml的二甲苯中,随后分别0.5mol和0.2mol异氰酸酯丙烯酸乙酯,在室温下反应2小时,得到丙烯酸酯封端的聚(二甲基硅氧烷)。在三颈瓶中分别加入0.05mol的aibn、4-氰基-4-[(十二烷基硫烷基硫羰基)硫烷基]戊酸和0.5mol的上述分子量为1000的丙烯酸酯封端的聚(二甲基硅氧烷)溶液,在氩气氛围下加热至80℃反应10小时,随后依次加入0.2mol的分子量为5000的丙烯酸酯封端的聚(二甲基硅氧烷)溶液和0.1mol的异氰酸酯丙烯酸乙酯,继续反应10小时,最终得到三嵌段的聚合物。
随后在三嵌段的聚合物加入0.05mol的六亚甲基二异氰酸酯进行交联,并加入0.05mol的硅氧烷交换催化剂:1,5,7-三氮杂二环[4.4.0]葵-5-烯/酮基布洛芬络合物,在60℃进行交联反应2小时,并在真空干燥箱内,80℃干燥24小时,得到交联的聚合物薄膜。
高分子网络拓扑结构异构过程:将聚合物薄膜加热至100℃,保持不同的反应时间。聚合物不同长度的侧链通过硅氧烷的可逆交换,交换后侧链分子量5000逐渐变小,1000的分子量逐渐变大,最后趋向平均。在交换后对材料的相关物性进行测试。例如:利用dsc测试材料玻璃化转变温度的变化,利用拉伸机测试模量和断裂伸长率的变化,利用afm测试材料的相分离程度。
实施例5
原料:
a)5-降冰片烯-2-甲醇:tci公司;结构式如下:
b)己内酯:tci公司;结构式如下:
c)六亚甲基二异氰酸酯,tci公司;其结构式如下:
d)辛酸亚锡:tci公司;
e)第二代grubbs催化剂:tci公司;
f)甲苯:阿拉丁(上海)试剂公司;
g)正己烷:阿拉丁(上海)试剂公司;
制备方法:
称取0.78mol的己内酯,0.1mol的降冰片烯-2-甲醇和0.5mmol辛酸亚锡,在氩气氛围下加热至120℃反应10小时。将所得到的产物溶解于50ml的甲苯后,再用正己烷沉淀得到液态的降冰片烯-2-甲醇-聚己内酯(分子量为1000da),产物置于于真空干燥箱内,80c干燥24小时。随后称取0.5mol的降冰片烯-2-甲醇-聚己内酯,和0.01mol第二代grubbs催化剂于三颈瓶中,在氩气氛围下加热至80℃反应2小时,随后加入0.5mol的降冰片烯-2-甲醇,继续80℃反应2小时,得到双嵌段的聚合物。
随后将双嵌段的聚合物溶解于甲苯中,加入0.05mol的六亚甲基二异氰酸酯,在60℃进行交联反应2小时,并在真空干燥箱内,80℃干燥24小时,得到交联的聚合物薄膜。
高分子网络拓扑结构异构过程:
将聚合物薄膜加热至40℃,保持不同的反应时间。聚合物的主链通过烯烃复分解由嵌段逐渐变成无规。在交换后对材料的相关物性进行测试。例如:利用dsc测试材料玻璃化转变温度的变化,利用拉伸机测试模量和断裂伸长率的变化,利用afm测试材料的相分离程度。
实施例6
原料:
a)乙酰乙酸甲基丙烯酸乙二醇酯:tci公司;结构式如下:
b)十八胺:tci公司;其结构式如下:
c)苄胺,tci公司;其结构式如下:
d)聚醚胺(d230):tci公司;
e)4-氰基-4-[(十二烷基硫烷基硫羰基)硫烷基]戊酸:tci公司;
f)偶氮二异丁腈(aibn):tci公司;
g)n,n-二甲基甲酰胺(dmf):阿拉丁(上海)试剂公司;
制备方法:
分别称取0.5mol十八胺和苄胺溶解于5ml的二甲苯中,随后分别0.5mol乙酰乙酸甲基丙烯酸乙二醇酯,在60℃下反应2小时,得到含二酮氨基动态共价键的甲基丙烯酸封端单体,并在并在真空干燥箱内,80℃干燥24小时。在三颈瓶中分别加入0.05mol的aibn、4-氰基-4-[(十二烷基硫烷基硫羰基)硫烷基]戊酸和0.5mol的上述十八胺制备的甲基丙烯酸封端单体,在氩气氛围下加热至80℃反应10小时,随后依次加入0.5mol的上述苄胺制备的甲基丙烯酸封端单体和0.05mol的乙酰乙酸甲基丙烯酸乙二醇酯,继续反应10小时,最终得到三嵌段的聚合物。
随后将三嵌段的聚合物溶解于10ml的dmf中,加入0.025mol的d230进行交联,在60℃进行交联反应2小时,并在真空干燥箱内,80℃干燥24小时,得到交联的聚合物薄膜。
高分子网络拓扑结构异构过程:将聚合物薄膜加热至120℃,保持不同的反应时间。嵌段聚合物的侧链通过转氨作用进行交换,交换后逐渐由嵌段变成无规。在交换后对材料的相关物性进行测试。例如:利用dsc测试材料熔融温度的变化,利用拉伸机测试模量和断裂伸长率的变化,利用afm测试材料的相分离程度。
实施例1制备的高分子网络拓扑异构体系在网络拓扑异构前后的熔点变化示意图如图1所示,实施例1在保持反应6小时后,其两个独立的熔点(约为95℃和40℃)合并为一个单一的熔点(68℃)其异构前后的应力应变曲线如图2所示(测试环境温度为80℃)。实施例2制备的高分子网络拓扑异构体系在不同光照时间下的熔点变化如图3所示,实施例2制备的高分子网络拓扑异构体系在不同光照时间下的模量和结晶度的变化如图4所示,实施例2制备的高分子网络拓扑异构体系在不同光照时间下的应力应变曲线如图5所示,实施例3制备的高分子网络拓扑异构体系在不同光照时间下的熔点变化如图6所示,实施例3制备的高分子网络拓扑异构体系在不同光照时间下的结晶度变化如图7所示,实施例4制备的高分子网络拓扑异构体系在异构前后的玻璃化转变温度变化如图8所示,应力应变曲线如图9所示,实施例5制备的高分子网络拓扑异构体系在异构前后的玻璃化转变温度变化如图10所示,实施例6制备的高分子网络拓扑异构体系在异构前后的玻璃化转变温度变化结果与图10相似。
下面以具体的应用例进一步说明本发明可以局部调控高分子材料物性的特点:
如图11所示,将实施例2所制备的高分子拓扑异构材料的三个区域分别在紫外光下照射0分钟,2分钟和5分钟,随后在80℃下保持2小时。由于光照时间不同,被照射区域的催化剂含量不同,因此在相同反应时间内的高分子异构化程度不同。对于实施例2而言,其催化剂含量越大,在相同时间内,侧链的链长越短,侧链密度越大,因此材料的熔点下降的越多。因此0分钟,2分钟和5分钟区域的熔点分别为50℃,40℃和30℃(此时结晶度已经很低,材料在室温下呈橡胶态)。因此在不同温度下(25℃,45℃,60℃),各个不同区域的材料会逐步从结晶态转变为橡胶态,在砝码的拉伸下从而被逐步拉长。该实施例很好的展示了该体系具有对高分子材料局部区域物性的调控能力,为构建非均质聚合物材料提供了新方法。
如图12所示,高分子网络会从嵌段型的网络结构(网络区域化学势相对较高)向混合型网络结构(网络区域化学势相对较低)演化为一种化学势驱动的网络拓扑异构。
当所述的网络热力学非稳态结构由网链构象数过低所造成时,高分子网络异构化过程如图13和14所示,分别为网链构象数驱动的网络拓扑异构示意图(重新分配链长型)和网链构象数驱动的网络拓扑异构示意图(改变网链结构型)。
以上所述的具体实施方式对本发明的技术方案和有益效果进行了详细说明,应理解的是以上所述仅为本发明的最优选实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充和等同替换等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!