一种砜吡草唑及其中间体的合成方法与流程
1.本发明涉及一种砜吡草唑及其中间体的合成方法,具体涉及一种利用新型中间体合成砜吡草唑的方法,属于砜吡草唑制备技术领域。
背景技术:
2.砜吡草唑(pyroxasulfone)是一种异噁唑类除草剂,由日本组合化学株式会社研究开发,化学名称为3-[5-(二氟甲氧基)-1-甲基-3-(三氟甲基)吡唑-4-基甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-异噁唑,分子式为c
12
h
14
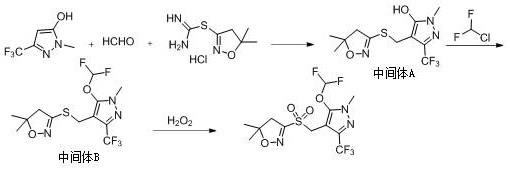
f5n3o4s,分子量为391.32, cas登录号为447399-55-5。砜吡草唑可用于大多数作物田的芽前土壤处理剂,施用后被杂草幼根与幼芽吸收,抑制幼苗早期生长,破坏分生组织与胚芽鞘,是植物体内vlcfa(极长侧链脂肪酸)(c20~c30)生物合成中严重的潜在抑制剂。其结构如下:目前,砜吡草唑的合成工艺报道的不多,专利cn102666502中公开了以下工艺路线:在碱性条件下,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛水溶液和5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐在水中进行缩合反应得到中间体a,中间体a再与二氟一氯甲烷烷基化得到中间体b,中间体b经双氧水氧化,得到砜吡草唑。
[0003]
上述工艺路线中,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛水溶液和5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐的反应过程中会产生大量的氨氮废水,这些废水难以处理,对环境和人类危害大。
技术实现要素:
[0004]
针对目前合成砜吡草唑中间体过程中存在的废水难以处理的不足,本发明提供了一种砜吡草唑中间体a的合成方法,该方法以s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯代替5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐来合成中间体a,反应快速、粗放,反应条件温和,不会产生难以处理的氨氮废水。
[0005]
本发明主要对背景中提到的中间体a的合成步骤进行了改进,将中间体5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐替换为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。本发明具体技术方案如下:一种式a所示的砜吡草唑中间体a的合成方法,所述中间体a由1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛和式(ⅲ)所示结构式的s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯在碱性水环境下反应得到,反应式如下:。
[0006]
本发明以s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯代替5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐与1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛反应合成中间体a,该反应为亲核取代反应,反应快速,无须催化剂的加入,在水环境下进行反应,反应副产物为乙酸盐,无氨氮废弃物,对人体和环境污染小,便于处理,避免了氨氮废水的产生和处理,大大简化了工艺流程。
[0007]
进一步的,中间体a在碱性水环境下进行,碱性环境有利于反应的进行。所述碱性环境可以由任意在水环境下使ph呈碱性的碱性物质提供。所述碱性物质可以是碱金属的氢氧化物,常用的为氢氧化钠或氢氧化钾。碱性物质的加入量为形成的酸的摩尔量的2倍或以上即可。
[0008]
进一步的,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛、s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯、碱性物质可以以纯物质的形式加入,也可以以水溶液的形式加入。它们的加入方式也没有严格的要求,可以一次性加入,也可以分批次的加入,也可以连续性的加入。
[0009]
进一步的,先将1-甲基-3-(三氟甲基)-1h-吡唑-5-醇和甲醛在碱性水环境下进行反应,然后再加入s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯进行反应,这样可以避免副反应的发生,提高原料的转化率。 1-甲基-3-(三氟甲基)-1h-吡唑-5-醇和甲醛发生的是羟甲基化反应,当检测到1-甲基-3-(三氟甲基)-1h-吡唑-5-醇没有或含量很低时,再加入s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯进行反应,当s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯反应完全时反应结束。可以通过hplc法来检测反应的进度。
[0010]
进一步的,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛、s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯的摩尔比为1:1-1.2:1-1.2,优选按照理论1:1:1的摩尔比进行。
[0011]
进一步的,水作为溶剂,其作用是为反应的进行提供良好的均相环境,其用量可以根据反应容器的利用率、反应效率、反应进行的困难度等情况进行选择。
[0012]
进一步的,中间体a的合成步骤简洁、反应条件温和,无须特殊催化剂和有机溶剂,仅在碱性水环境中进行反应即可。反应温度一般为10-40℃,反应温度较低,能耗低。该反应温度指的是1-甲基-3-(三氟甲基)-1h-吡唑-5-醇和甲醛的反应温度,以及加入s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯后的反应温度。
[0013]
进一步的,反应后的反应液经调整ph至中性、析晶、过滤、干燥的操作,得到中间体a。ph可以通过盐酸等酸来调整。该后处理过程简洁、粗放,产品容易分离,分离产品后的母
液中含有乙酸盐副产物,不含氨氮废物,更易于处理。
[0014]
进一步的,本发明所用的1-甲基-3-(三氟甲基)-1h-吡唑-5-醇可以根据现有技术中报道的方法进行制备,例如按照文献:journal of agricultural and food chemistry (2008), 56(22), 10805-10810中公开的方法进行制备。
[0015]
进一步的,本发明所用的s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯是一种新的化合物,其结构式如下式(ⅲ)所示。该化合物可以通过式(i)所示的化合物(3-卤代-5,5-二甲基-4,5-二氢异恶唑)与式(ⅱ)所示的硫代乙酸钾反应得到,反应式如下:。
[0016]
上述式(i)中,x为离去基团,其可以是与式(ⅱ)的化合物接触后容易离去的基团,例如氯、溴、碘、otf、oms、ots、ons等,其中,oms为甲基磺酰氧基、ots为对甲苯磺酰氧基、otf为三氟甲磺酰氧基、ons为对硝基苯磺酰氧基。otf、oms、ots、ons具体结构式下:。
[0017]
s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯的合成反应条件温和、简单、高效,无须特殊催化剂,无需调整ph。两反应原料在反应溶剂以及合适的反应温度下即可发生反应。所用的反应溶剂是为原料的反应提供合适的反应环境,反应溶剂选择有机的、不与原料与产物反应、能够很好的提供均相反应环境的溶剂即可,例如在有机反应中经常作为溶剂使用的醇类溶剂、腈类溶剂、酮类溶剂、酰胺类溶剂等,包括但不限于乙醇、乙腈、甲醇、丙酮、n,n-二甲基甲酰胺。反应溶剂能保证反应良好的进行即可,其使用量可以根据反应容器的利用率、反应效率、反应进行的困难度等情况进行选择。
[0018]
进一步的,s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯合成的反应温度为50℃~80℃。在此温度内,所得产品收率较高。为了使反应更好的进行,优选在搅拌下进行反应。
[0019]
进一步的,上述式(i)所示的化合物中,当x为氯、溴、碘时,可以按照文献:journal of agricultural and food chemistry (2008), 56(22), 10805-10810、专利cn101389625a中公开的方法进行制备,当x为otf、oms、ots、ons时,可以由5,5-二甲基-4,5-二氢异噁唑-3-酮与取代的磺酰氯(rx)反应得到,反应式如下:其中,5,5-二甲基-4,5-二
氢异噁唑-3-酮在cn101389625a、cn109574945a中有报道,可以按照这些现有技术中报道的方法制备。5,5-二甲基-4,5-二氢异噁唑-3-酮与取代的磺酰氯(rx)可以按照现有技术中报道的步骤和工艺条件进行反应。
[0020]
上述硫代乙酸钾可以通过市购的方式得到。式(i)所示的化合物与式(ⅱ)所示的硫代乙酸钾的摩尔比为1:1.05~1.2。式(i)所示的化合物、硫代乙酸钾以及反应溶剂可以按照任意的顺序进行混合,反应原料可以一次性加入,为了更好的混合均匀,也可以分批次或者连续的加入,反应原料可以直接加入,也可以先用反应溶剂溶解后以溶液的形式加入。
[0021]
进一步的,s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯合成时,通过hplc法来检测反应的进度,当检测到没有式(i)所示的化合物时反应结束。反应后的反应液冷却至室温,过滤除去无机盐,滤液回收溶剂,得s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯油状物产物。该后处理过程简洁、粗放,产品和废盐容易分离,溶剂可以回收循环利用,不会产生废水。
[0022]
进一步的,本发明还提供了一种砜吡草唑的合成方法,该方法包括按照上述方法合成中间体a的步骤。
[0023]
进一步的,上述合成砜吡草唑的方法中,除了合成中间体a的步骤外,还包括中间体a与二氟一氯甲烷进行烷基化反应得到中间体b、以及中间体b与双氧水进行氧化反应得到砜吡草唑的步骤。整个反应过程的工艺路线如下:。
[0024]
进一步的,合成中间体b以及由中间体b合成砜吡草唑的具体工艺过程可以按照现有技术中报道的方法进行,例如可以按照专利cn102666502a中描述的方式进行,这一部分属于现有技术,在此不再赘述。
[0025]
在本发明某一具体实施例中,中间体a与二氟一氯甲烷的反应在有机溶剂和缚酸剂存在下进行,缚酸剂为现有技术中报道的任意能够将反应形成的酸消耗掉的物质,常用的为碳酸钠、碳酸钾、三乙胺。有机溶剂为现有技术中报道的可行的有机溶剂,例如乙腈等。
[0026]
在本发明某一具体实施例中,中间体a与二氟一氯甲烷的摩尔比为1:1.2-3,反应温度为10℃~30℃,反应过程中时时通过hplc检测中间体a的含量,当中间体a完全反应时反应结束。反应结束后,过滤去除副产物盐,反应液无须提取产品直接可以用于下一步反应。
[0027]
在本发明某一具体实施例中,中间体b与过氧化氢的摩尔比为1:5~10,中间体b与双氧水的反应温度为60℃-80℃。双氧水的浓度可以随意选择,一般选择较高浓度。反应过程中时时通过hplc检测中间体b含量,当中间体b完全反应时反应结束。反应结束后,减压蒸馏去除有机溶剂,然后过滤产生的固体,即为砜吡草唑。
[0028]
本发明的优点在于:(1)优化了砜吡草唑的反应路线,以s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙
酯代替5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐,减少了中间体a合成过程中氨氮废水的产生,降低了废水的处理难度和对人体和环境的污染。
[0029]
(2)s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯和中间体a的合成具有反应速度快、反应条件温和、不需要特殊的催化剂、不需要过高的温度和高压、对设备要求均较低等优势,具有很好的工业化应用价值。
[0030]
(3)整个合成过程和后处理过程简单粗放,反应得到的产物容易分离、方便处理,简化了生产工艺,适合工业化大生产。
[0031]
(4)整个反应选择性高、反应收率较高、纯度较高,砜吡草唑收率在81%以上。
具体实施方式
[0032]
以下结合具体实施例对本发明做进一步说明。下述说明仅是示例性的,并不对保护范围进行限制。在不脱离本发明发明构思的基础上,本领域技术人员不经创造性劳动所得的其他实施方案,也在保护范围之内。
[0033]
下述实施例中,如无特别说明,所述浓度均为质量百分浓度。
[0034]
下述实施例中,所用原料如无特别说明,均为市购产品。
[0035]
下述实施例中,收率=产品的实际质量
×
纯度/产品的理论质量。
[0036]
(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯的合成实施例1室温下,在60ml乙醇中依次加入3-氯-5,5-二甲基-4,5-二氢异恶唑(式(i)所示的化合物)13.3g和硫代乙酸钾12.1g,然后加热至80℃搅拌反应,4hr后用hplc检测无3-氯-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除乙醇,恒重后得到油状物17.7g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为93%,收率以3-氯-5,5-二甲基-4,5-二氢异恶唑计为95.1%。
[0037]
实施例2室温下,在60ml乙腈中依次加入3-氯-5,5-二甲基-4,5-二氢异恶唑13.3g和硫代乙酸钾13.6g,然后加热至75℃搅拌反应,4hr后用hplc检测无3-氯-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除乙腈,恒重后得到油状物17.9g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为94%,收率以3-氯-5,5-二甲基-4,5-二氢异恶唑计为97.3%。
[0038]
实施例3室温下,在60ml甲醇中依次加入3-氯-5,5-二甲基-4,5-二氢异恶唑13.3g和硫代乙酸钾12.7g,然后加热至65℃搅拌反应,5hr后用hplc检测无3-氯-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除甲醇,恒重后得到油状物17.2g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为94%,收率以3-氯-5,5-二甲基-4,5-二氢异恶唑计为93.5%。
[0039]
实施例4室温下,在60mln、n二甲基甲酰胺中依次加入3-氯-5,5-二甲基-4,5-二氢异恶唑13.3g和硫代乙酸钾12.6g,然后加热至80℃搅拌反应,6hr后用hplc检测无3-氯-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除n、n
二甲基甲酰胺,恒重后得到油状物17.5g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为93%,收率以3-氯-5,5-二甲基-4,5-二氢异恶唑计为94.1%。
[0040]
实施例5室温下,在60ml丙酮中依次加入3-甲磺酰氧基-5,5-二甲基-4,5-二氢异恶唑19.3g和硫代乙酸钾12.6g,然后加热至60℃搅拌反应,6hr后用hplc检测无3-甲磺酰氧基-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除丙酮,恒重后得到油状物17.2g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为95%,收率以3-甲磺酰氧基-5,5-二甲基-4,5-二氢异恶唑计为94.4%。
[0041]
实施例6室温下,在60ml丙酮中依次加入3-三氟甲磺酰氧基-5,5-二甲基-4,5-二氢异恶唑24.7g和硫代乙酸钾12.6g,然后加热至60℃搅拌反应,6hr后用hplc检测无3-三氟甲磺酰氧基-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除丙酮,恒重后得到油状物17.4g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为94%,收率以3-三氟甲磺酰氧基-5,5-二甲基-4,5-二氢异恶唑计为94.5%。
[0042]
实施例7室温下,在60ml丙酮中依次加入3-对甲苯磺酰氧基-5,5-二甲基-4,5-二氢异恶唑26.9g和硫代乙酸钾12.6g,然后加热至60℃搅拌反应,6hr后用hplc检测无3-对甲苯磺酰氧基-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除丙酮,恒重后得到油状物17.4g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为94%,收率以3-对甲苯磺酰氧基-5,5-二甲基-4,5-二氢异恶唑计为94.5%。
[0043]
实施例8室温下,在60ml丙酮中依次加入3-对硝基苯磺酰氧基-5,5-二甲基-4,5-二氢异恶唑30.1g和硫代乙酸钾12.6g,然后加热至60℃搅拌反应,6hr后用hplc检测无3-对硝基苯磺酰氧基-5,5-二甲基-4,5-二氢异恶唑,反应结束,反应液冷却至室温,过滤除去无机盐,过滤后的母液减压蒸除丙酮,恒重后得到油状物17.4g,即为s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯。经hplc检测,产品纯度为94%,收率以3-对硝基苯磺酰氧基-5,5-二甲基-4,5-二氢异恶唑计为94.5%。
[0044]
砜吡草唑的合成实施例91、中间体a合成:室温下,在40ml水中依次加入1-甲基-3-(三氟甲基)-1h-吡唑-5-醇16.6g、37wt%甲醛水溶液8.1g和氢氧化钠8.3g,搅拌2hr后,在10℃加入s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯17.3g,然后在此温度下继续反应,继续反应7hr后hplc检测显示1-甲基-3-(三氟甲基)-1h-吡唑-5-醇完全反应,反应结束后将反应液加入37wt%盐酸20.5g,然后搅拌析晶,抽滤得白色固体沉淀,干燥后得28.8g固体,即为中间体a,经hplc检测纯度为99%,以
1-甲基-3-(三氟甲基)-1h-吡唑-5-醇计收率为92.3%。
[0045]
2、中间体b合成:向40ml乙腈中加入6.9g碳酸钾和14.4g中间体a,20℃搅拌2hr,常压下缓缓通入二氟一氯甲烷气体8.5g,8hr后hplc检测中间体a反应完全,反应结束后过滤除去固体,母液直接用来下一步反应。
[0046]
3、砜吡草唑合成:室温下往上述步骤2的反应液中加入30wt%双氧水28.3g,搅拌1hr后,缓缓升温至60℃进行反应,4hr后hplc检测显示反应结束。减压浓缩除去乙腈后,反应液冷却至室温,过滤得白色固体16.8g,即为砜吡草唑,经hplc检测其含量为98.4%,以中间体a计,砜吡草唑收率为84.5%。
[0047]
实施例101、中间体a合成:室温下,在80ml水中依次加入1-甲基-3-(三氟甲基)-1h-吡唑-5-醇33.2g、37wt%甲醛水溶液16.2g和氢氧化钠16.6g,搅拌2hr后,在30℃加入s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯34.6g,然后在此温度下继续反应,继续3hr后hplc检测反应结束,反应液加入37wt%盐酸41g,搅拌析晶,抽滤得白色固体沉淀,干燥后得57.5g固体,即为中间体a,经hplc检测纯度为99%,以1-甲基-3-(三氟甲基)-1h-吡唑-5-醇计收率为92.1%。
[0048]
2、中间体b合成:向40ml乙腈中加入10.6g碳酸钠和30.9g 中间体a,20℃搅拌2hr,常压下缓缓通入二氟一氯甲烷气体15g,8hr后hplc检测中间体a完全反应,反应结束后过滤除去固体,母液直接用来下一步反应。
[0049]
3、砜吡草唑合成:室温下往上述步骤2的反应液中加入30wt%的双氧水113.3g,搅拌1hr后,缓缓升温至60℃进行反应,4hr后hplc检测显示反应结束。减压浓缩除去乙腈后,冷却至室温,过滤得白色固体34.1g,即为砜吡草唑,经hplc检测其含量为98.1%,以中间体a计,砜吡草唑收率为87.5%。
[0050]
实施例111、中间体a合成:室温下,在80ml水中依次加入1-甲基-3-(三氟甲基)-1h-吡唑-5-醇33.2g、37wt%甲醛水溶液16.2g和氢氧化钠16.6g,搅拌2hr后,在40℃加入s-(5,5-二甲基-4,5-二氢异恶唑-3-基)乙硫酸乙酯34.6g,然后在此温度下继续反应,继续反应2hr后hplc检测1-甲基-3-(三氟甲基)-1h-吡唑-5-醇反应完全,反应结束后反应液加入37wt%盐酸41g,搅拌析晶,抽滤得白色固体沉淀,干燥后得56.8g固体,即为中间体a。经hplc检测纯度为99%,以1-甲基-3-(三氟甲基)-1h-吡唑-5-醇计收率为91.0%。
[0051]
2、中间体b合成:向40ml乙腈中加入10.6g碳酸钠和30.9g 中间体a,20℃搅拌2hr,常压下缓缓通入二氟一氯甲烷气体16g,8hr后hplc检测中间体a反应完全,反应结束后过滤除去固体,母液直接用来下一步反应。
[0052]
3、砜吡草唑合成:
室温下往上述步骤2的反应液中加入30wt%的双氧水56.6g,搅拌1hr后,缓缓升温至80℃进行反应,4hr后hplc检测显示反应结束。减压浓缩除去乙腈后,冷却至室温,过滤得白色固体33.1g,即为砜吡草唑,经hplc检测其含量为98.7%,以中间体a计,砜吡草唑收率为83.5%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1